
From cancer and autoimmune diseases to allergies and neurodegenerative diseases, antibody therapeutics are being used to treat human conditions. Molecular properties of antibodies, such as their high affinities, long half-lives, potent effector functions, and excellent biophysical properties (i.e., high stability and solubility), contribute significantly to the success of antibody therapeutics.
Antigen-specific monoclonal antibodies (mAbs) may be designed computationally using generative machine learning (ML). Unfortunately, confirmation of this hypothesis has been impeded by the infeasibility of testing large numbers of antibodies on their most critical design attributes: paratopes, epitopes, affinity, and developability. It is currently not common for mAbs to be developed based largely on rule-driven discovery but rather on screening libraries and heuristics, with very little to no emphasis placed on rule-driven discovery.
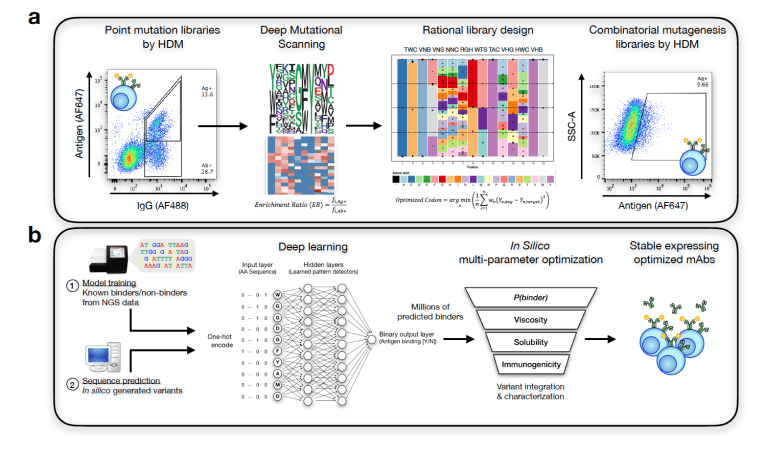
Image Source: doi: https://doi.org/10.1101/617860
A long-standing approach to protein engineering involves using machine learning models to guide protein synthesis based on data generated by mutagenesis libraries. In directed evolution, DNA, RNA, and protein sequences are linked to phenotypic outputs, making them well-suited for this kind of analysis. Molecular phenotypes can be predicted from sequence data using deep learning models developed in recent years by means of deep sequencing and parallel computing. An example of a large-scale, high-dimensional data set is the millions of reads acquired from a single deep sequencing experiment, which uses deep learning to uncover the relationship between them. Since model extrapolation allows interrogation of a much larger sequence space than what is physically possible, it is particularly useful for protein engineering.
By showing (in a 3D-lattice space) that sufficiently large training data can be used to generate antibody variants with high affinity and specific epitope binding, deep learning can learn the non-linear rules of 3D antibody-antigen interaction. The use of machine learning for sequence generation also helped discover novel parameter combinations for developability. However, some deep learning methods reproduce the training data with minimal changes, which Renz and colleagues call the “copy problem .”When molecular properties (in our case, antigen binding and developability) cannot be tested high-throughput, the copy problem is particularly prevalent. Because prospective testing is not available, the generated dataset cannot be functionally evaluated (e.g., antigen binding), making addressing the copy problem somewhat impossible (testing sequence diversity on unknown binding modes does not provide insight into diversity).
Many of the antibody candidates that are selected from in vitro library sorting or immunization usually have a variety of biophysical properties in common formulation conditions, including various solubilities and viscosities. The production, formulation, and/or delivery of antibody candidates with the highest bioactivities is often hindered by their undesirable biophysical properties. As a result of this discovery, potentially promising candidates can be compromised later in the development process after a substantial investment of limited resources. Thus, antibody engineering techniques are needed to improve their biophysical properties while preserving their high affinity and bioactivity, particularly during development.
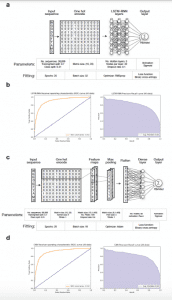
Image Source: doi: https://doi.org/10.1101/617860
It will be crucial to develop simple and robust methods that are co-optimized in terms of antibody affinity and biophysical properties will be of great importance for predicted CDR mutations. Through a combination of display technologies (e.g., phage and yeast-surface display) and deep sequencing of antibodies, large datasets have been generated that correlate multisite antibody mutations with different levels of properties (e.g., high and low levels of affinity, stability, or specificity). Using frequencies or enrichment ratios to identify promising antibody mutations in deep sequencing datasets is a common method of analysis, but these approaches fail to take advantage of a large amount of sequence information and, in some cases, only lead to moderate improvement in antibody properties.
It is logical and simple to encode the amino acid sequences of antibody libraries into sequence identity vectors (e.g., one-hot encoded antibody sequences) in order to build predictive models based on the vast amounts of sequence information obtained from deep sequencing antibody libraries. These models often provide accurate predictions of antibody properties, such as high or low affinity, when developed using this approach.
For treating non-small cell lung cancer, the drug c-Met [also known as tyrosine-protein kinase Met or hepatocyte growth factor receptor (HGFR)] reached phase II clinical trials. Emibetuzumab’s nonspecific binding has been attributed primarily to its heavy chain CDR in previous research. Based on the evidence that heavy chain CDRs play an important role in determining antibody affinity, it was expected mutations in the heavy chain of emibetuzumab to have strong effects on on-target (affinity) and off-target (nonspecific) binding. In LDA models, one-dimensional projections show that biophysical properties are continuously variable. Using these projections, which are linear combinations of learned scalings that maximize interclass separation, antibodies that are Pareto optimal can be directly identified despite being trained on binary datasets for affinity and specificity.
Drug development is impeded by the difficulty and time-consuming process of co-optimizing multiple antibody properties. In spite of the small number of libraries screened and deep sequenced, the researchers were able to train deep neural networks that were capable of accurately predicting antigen-binding through the analysis of antibody sequences. The use of deep learning models enables the prediction of novel antibody mutations that optimize affinity and specificity beyond the capabilities of the original antibody library. As a result of these findings, machine learning models can expand the exploration of antibody sequence space and accelerate the development of highly potent, drug-like antibodies.
Story Sources:
Makowski, E.K., Kinnunen, P.C., Huang, J. et al. Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space. Nat Commun 13, 3788 (2022). https://doi.org/10.1038/s41467-022-31457-3.
R. Akbar et al., “In silico proof of principle of machine learning-based antibody design at unconstrained scale,” MAbs, vol. 14, no. 1, p. 2031482, 2022.
D. M. Mason et al., “Deep learning enables therapeutic antibody optimization in mammalian cells by deciphering high-dimensional protein sequence space,” bioRxiv, p. 617860, 2019.
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.











{kind=link}