

In the pursuit of understanding how RNA structure and function are related, predicting RNA structures becomes a tool alongside experimental approaches. However, the limited availability of labeled training data poses a challenge for learning in this field. To address this challenge, Scientists from Germany introduced BARNACLE, a method that combines self-supervised pre-training with XGBoost modeling to enhance RNA contact prediction. This approach shows improvements compared to existing methods, highlighting its potential for studying RNA and other fields in the natural sciences that lack labeled data.
At the molecular level, our lives are orchestrated by interactions between biomolecules. Understanding their structures is essential for gaining insights into their functions. Despite advancements in protein structure prediction, RNA, a vital biomolecule with diverse functions, has received less attention. It plays a crucial role in the biology of all living organisms and is considered one of the three major macromolecules, along with DNA (Deoxyribonucleic Acid) and proteins.
RNA, comprising three types viz. messenger RNA (mRNA), transfer RNA (tRNA), and ribosomal RNA (rRNA), plays vital roles in biological processes. Messenger RNA (mRNA) conveys genetic information for protein synthesis, constituting 5% of total RNA. Transfer RNA (tRNA), which makes up 15% of mRNA, carries amino acids based on mRNA codons. Ribosomal RNA (rRNA) forms ribosomes, which are essential for protein assembly and catalyzing biochemical reactions, highlighting RNA’s therapeutic potential in treating diseases. It is speculated that RNA may have existed before DNA developed in the earliest phases of life, indicating a crucial role for RNA at the beginning of life. One key to making new discoveries in life science is predicting the structure of RNA.
Key Obstacles in Predicting RNA Structure
RNA molecules can fold into one or more competing three-dimensional (3D) structures similar to proteins since they are made of linear ribonucleotide strands. However, due to the scarcity of high-quality annotated data, predicting RNA structures is challenging. Such information is currently in short supply for RNA but abundant for proteins. Due to the lack of available data, BARNACLE concentrates on predicting spatial relationships, or “contact maps,” which serve as a stand-in for three-dimensional (3D) RNA structures. But BARNACLE has XGBoost, a combination of self-supervised pre-training and XGBoost modeling.
The Power of XGBoost Modeling
A key component of BARNACLE is XGBoost, a machine learning technique renowned for its effectiveness and robustness. It effectively uses sparsely labeled data, surpassing deep neural networks and known baselines. The mixture of self-supervised pre-training and XGBoost modeling results in substantial improvements in RNA contact prediction.
RNA Contact Prediction Through BARNACLE
The RNA contact prediction process encompasses self-supervised pre-training and downstream training for RNA contact prediction.
Self-Supervised Pre-Training
Self-supervised pre-training involves several tasks, including inpainting, jigsaw, bootstrapping, and contrastive augmentations. Each task serves as a pretext task to learn representations and patterns beneficial for the downstream task. These tasks are chosen based on their compatibility and effectiveness in the context of RNA data.
Inpainting: Similar to masked language modeling, it involves token masking and subsequent recovery. Key hyperparameters include the fraction of masked tokens, token replacement strategies, and mask shapes. For the final model, individual tokens are randomly replaced with a random legal token. Inpainting training tends to converge slowly, possibly due to random mask generation.
Jigsaw: It entails splitting RNA sequences into chunks and shuffling them based on assigned permutations. Permutations can be applied per sequence or across the entire MSA (Multiple Sequence Alignment). Key parameters include the number of chunks and permitted permutations. The model’s goal is to predict the applied permutation through multi-class classification. Jigsaw tasks converge faster but do not consistently enhance downstream performance.
Bootstrapping: This task involves generating synthetic sequences based on position-wise token frequency distributions in the MSA. This can replace entire sequences or operate at the token level. The model must distinguish between original and replaced sequences/tokens, creating a binary classification problem.
Contrastive: Unlike the other upstream tasks, it does not require any augmentation. Instead, for each sequence in each MSA of the current batch, the model produces a separate latent vector. Then, it tries to make the data within the same group look similar to each other (the cosine similarity between sequences of the same MSA is maximized) while minimizing sequences from different MSAs.
Downstream Training
Two models are employed for downstream contact prediction: logistic regression and boosted decision trees (XGBoost).
- Logistic Regression: This classic approach models the log-odds of a residue pair in contact as a linear combination of attention map entries. Log-odds are transformed into contact probabilities using the sigmoid function.
- Boosted Decision Trees: Gradient-boosted trees (XGBoost) are used as ensemble models. Each tree aims to reduce the deficiency of the previous model concerning training data, as measured by a differentiable loss function. The model score is interpreted as log-odds, with the sigmoid function applied for classification.
Fine-tuning involves optimizing the pre-trained backbone for the downstream task and data. This can be done with either the logistic regression model or XGBoost. For the logistic regression model, only the parameters of the added downstream model are optimized, while for XGBoost, both the backbone and downstream model parameters are optimized. Early stopping based on downstream metrics is used to prevent overfitting. The fine-tuned regression model is complete at this point, while the fine-tuned XGBoost model uses the re-trained backbone from the fine-tuned regression model.
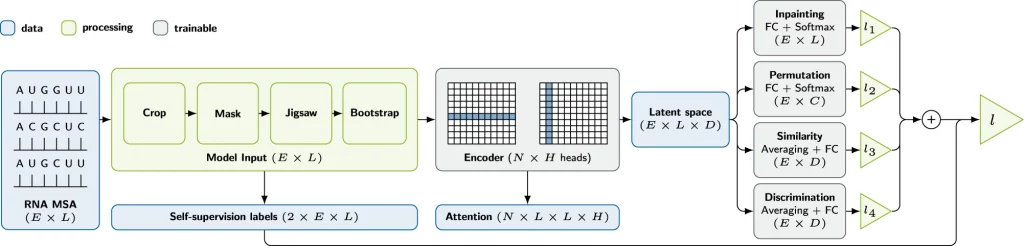
Image Source: https://doi.org/10.1038/s42003-023-05244-9
What BARNACLE Brings to the Table
BARNACLE’s performance was compared to the other RNA contact prediction tools, such as CoCoNet, and BARNACLE was found to be better. In the context of predicting the three-dimensional structure of RNA, “contacts” refer to pairs of nucleotides that are geographically adjacent to or interact with one another. Understanding the RNA’s overall folding and structure, which in turn offer insights into its biological function, depends on predicting these connections. BARNACLE has a much higher top-L precision than CoCoNet, with 81% compared to 47% for CoCoNet.
Taking into account true positives, true negatives, false positives, and false negatives, the MCC (Matthews Correlation Coefficient) is a thorough metric. It quantifies the model’s overall effectiveness. While the baseline (a reference point) also has a 25% MCC, BARNACLE obtains a 25% MCC. This shows that BARNACLE outperforms the starting point significantly. BARNACLE’s MCC is higher at 25% compared to the baseline’s 25%. This is all thanks to fine-tuning the model and integrating it with XGBoost, which helps it outperform CoCoNet and almost double its MCC.
Conclusion
BARNACLE is a revolutionary approach to RNA contact prediction that can be used to bridge the gap between known sequences of RNA and those experimentally resolved in tertiary structures. This approach can potentially address data scarcity in various natural sciences disciplines, and its adaptability to downstream applications highlights the need for resource-intensive model training. The selection of a suitable downstream metric remains a key factor in evaluating BARNACLE. It also highlights the flexibility of this model approach for downstream applications, which reduces the need for resource-intensive model training. The selection of the appropriate downstream metric remains critical, as different metrics influence performance measurement.
Article Source: Reference Article
Learn More:




Prachi is an enthusiastic M.Tech Biotechnology student with a strong passion for merging technology and biology. This journey has propelled her into the captivating realm of Bioinformatics. She aspires to integrate her engineering prowess with a profound interest in biotechnology, aiming to connect academic and real-world knowledge in the field of Bioinformatics.





{kind=link}