

Researchers from the University of Oslo have developed a novel genotyper named KAGE for detecting SNPs, short insertions, and deletions. KAGE uses an alignment-free word-based approach to genotype variants in a shorter duration, higher accuracy, and less memory.
Genotyping refers to the process of detecting the genotype (genetic makeup) of an individual using biological techniques such as RAPD (Random Amplification of Polymorphic DNA), RFLP (Restriction Fragment Length Polymorphism), PCR (Polymerase Chain Reaction), and microarrays. The individuals’ genotype is compared to a reference sequence or another sample to identify small differences such as SNPs (Single Nucleotide Polymorphism) and indels. The 1000 genomes project is a collaborative plan among researchers from the US, UK, Germany, and China to sequence and put forth a catalog of genetic variants (such as structural variants, SNPs, and haplotypes) from over 1000 individuals across the globe. This comprehensive project has enabled researchers to access information on different variant sites from a sub-population to an individual level.
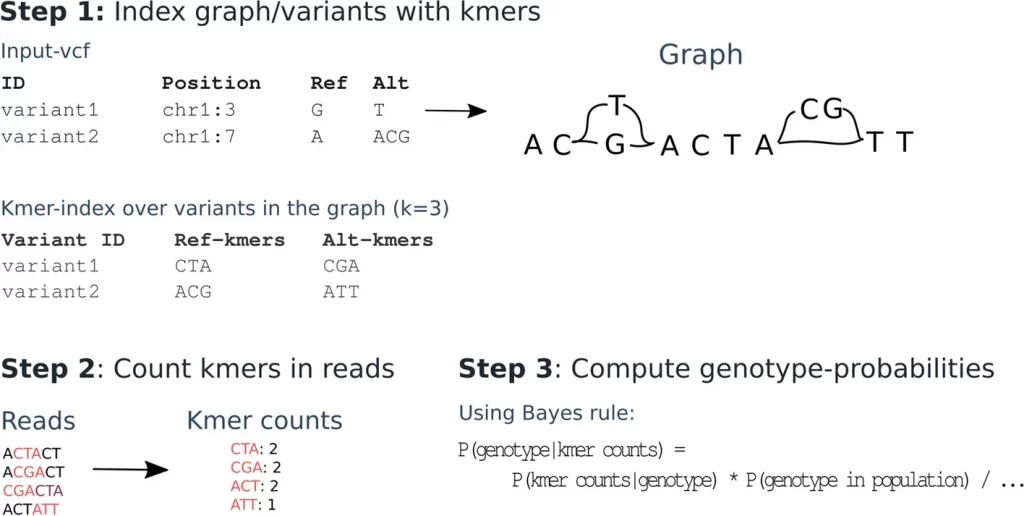
Image Source: https://doi.org/10.1186/s13059-022-02771-2
The genetic variations are found and characterized in two steps,
1) Genetic variation discovery (also termed variant calling) – The individuals’ genome is sequenced, aligned, and mapped with a reference genome to identify variants.
2) Genotyping – The identified variations are probed for their presence in one or both chromosomes in the queried genome.
Genotyping is traditionally performed using SNP chips or sequencing. While these methods show accuracy, they are slow in generating large-scale results. To avoid this drawback, alignment-free approaches have been developed for the past few years. Alignment-free techniques can be divided into two categories,
1) Word-based methods
2) Information theory-based methods
In word-based methods, the words or k-mers (substrings with the length of k) surrounding the variations are probed. However, this method is also faulty as the alleles of the variations do not share unique k-mers. MALVA is the first mapping-free genotyping tool that solves the issue by taking longer k-mers. Another approach, PanGenie, supersedes MALVA by combining haplotype information to detect variants and genotypes and does not have unique k-mers using a Hidden Markov Model (HMM). But PanGenie cannot assess the large variant and haplotype information available in the 1000 Genomes Project. And the information theory-based approach work by recognizing and calculating the information shared by the two biological sequences. Here, nucleotides and amino acids are represented as strings which are then recognizable by information theory tools based on their complexity.
Therefore, to address these drawbacks in word-based methods, Grytten et al. have introduced KAGE – a novel genotyper that detects SNPs and indels using an alignment-free approach to improve efficiency and utilizing haplotype information for improved accuracy.
Findings
KAGE outperformed other alignment-free genotypers by completing an entire sample in 12 minutes and 16 computer cores. PanGenie produced accurate results on indels but failed to determine SNPs accurately. PanGenie also took 20 times longer run-time ( approximately 240 minutes or 4 hours) and more memory than KAGE. MALVA performed poorly in detecting both SNPs and indels. These results imply that KAGE is suitable to genotype any sample using a standard laptop in any clinical space without the requirement of any high-end equipment.
GATK HaplotypeCaller was used to identify any potential variants from the reference genome, resulting in higher accuracy of genotype identification than previous benchmark methods such as GraphTyper and MALVA. The authors were astonished to find that KAGE and PanGenie showed an ability to identify variants with high accuracy, similar to GraphTyper, the traditional benchmark. GraphTyper is a graph-based variant caller where the query sequence is mapped, aligned, and realigned with the sequence graph. But the fact that KAGE and PanGenie can achieve such accuracy without any alignment requirements represents seminal progress in genotyping. PanGenie, despite the high precision, has a slow and memory-consuming performance due to the complex Hidden Markov Model, making it a less suitable option.
Imputating KAGE with other sophisticated tools such as GLIMPSE or any other tool the user wishes seems to work well and brings higher accuracy. On the other hand, MALVA can be run at a considerably lower run time just by optimizing it for multiple computer cores.
Limitations
1) KAGE can only genotype SNPs, short insertions, and deletions, which is compared with tools for SNPs and indels only. KAGE is not yet optimized for genotyping structural variations, while several methods perform those as well.
2) KAGE relies heavily on a population variation database such as the 1000 Genomes Project, but it ignores the fact that not all individuals and variants are represented in the pangenome databases.
3) KAGE will not work on other species whose pangenomes are not available yet.
4) KAGE and other word-based methods cannot use information from paired-end reads, where the sequence is read from both ends, thereby generating high-quality alignable data. But the authors do not see this as a serious disadvantage, as single-end sequencing commonly opts for its low cost and prevalence.
Conclusion
Genotyping is crucial to mapping out deleterious phenotypes and disease associations and is also important to understand evolution. With millions of SNPs identified in the human genome resulting in traits such as eye color and not-so-desirable genetic diseases like sickle cell anemia, cystic fibrosis, etc., identifying genotypes from a query sequence with high accuracy in a shorter run time is of high importance. KAGE is a notable contribution to the field as it allows the detection of SNPs, short insertions, and deletions using an alignment-free approach with a significantly faster run time than its competitors. Researchers can use KAGE to genotype many genomes, such as detecting variants in samples from the 100,000 genomes project in the near future.
Article Source: Reference Paper | Datasets Analyzed: GitHub | An index to be used with KAGE: Zonodo| KAGE: GitHub | Zenodo
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}