
Modeling of protein-small molecule systems offers advantages in speed and generality for exploring interactions with small molecules in the folded state; the modeling of conformational heterogeneity in protein-small molecule systems is deemed an outstanding issue at the atomic level. Researchers from the University of Washington created a graph neural network named ChemNet that was taught to reproduce accurate atomic positions from partially damaged input structures from the Protein Data Bank and the Cambridge Structural Database. ChemNet is a quick and random tool that reliably creates small molecule structures according to their bonding and atom composition. Moreover, it describes the broader protein context, which makes protein-small molecule docking possible.
Introduction
Atomistic modeling of interactions between metals, nucleic acids, small chemical and inorganic compounds, and proteins is still a complex topic. Although DiffDock, a deep learning-based small molecule docking tool, outperforms previous approaches like Glide and GNINA/SMINA, it is less effective on unknown receptors and in high accuracy regimes. Best-in-class results have been demonstrated by DiffDock and Torsional Diffusion on the synthetic GEOM-DRUGS dataset of drug-like compounds. Unfortunately, the capacity to simulate the entire range of protein functions is limited by the fact that these methods only model particular groups of interaction partners. With evolutionary-related protein sequences and structures, RoseTTAFold (RF) and AlphaFold2 (AF2) have made it possible to predict the atomic structure of proteins and protein-protein complexes. Lately, these techniques have been expanded to include the modeling of more general biomolecular systems, including protein-nucleic acid complexes. Even while AlphaFold3 (AF3) seems to perform better than other techniques, it is not yet generally accessible and may not be able to simulate conformational heterogeneity.
Advantages of ChemNet
- ChemNet’s stochastic and fast processing speed enables mapping conformational heterogeneity by creating prediction ensembles.
- Higher success rates and activities are achieved in enzyme design endeavors when ChemNet is utilized to evaluate the precision and pre-organization of active sites. The fast generation of conformational ensembles of small molecules and small molecule protein systems is anticipated to be a benefit of ChemNet.
Diving Deep into the intricacies of ChemNet
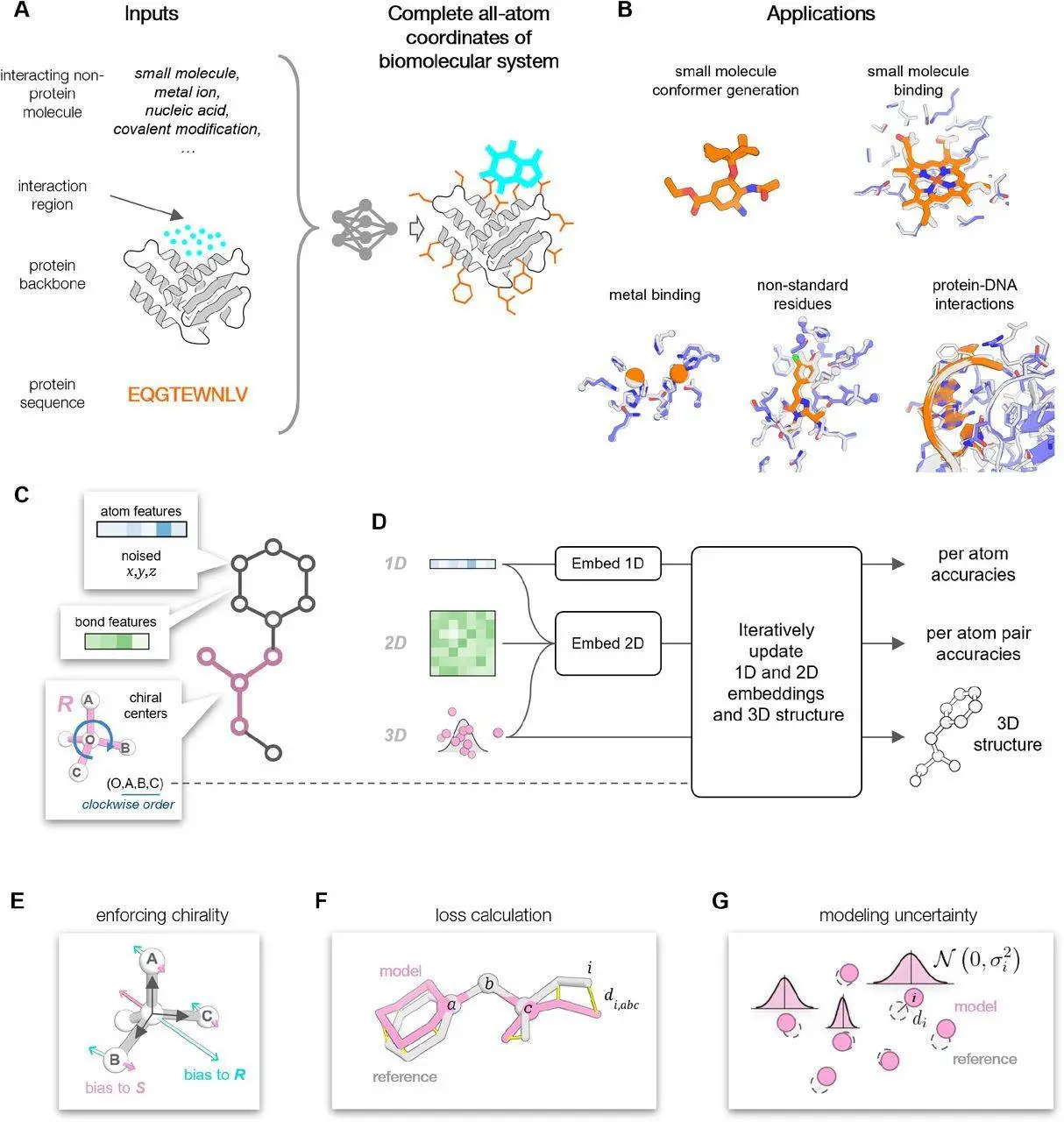
Researchers created the stochastic deep neural network known as ChemNet to mimic small compounds and their interactions with proteins atomistically. The task is to dock a small molecule into the region of interest while adjusting the conformations of the protein side chains and the molecule itself. In many applications, a reliable structure of the target protein can be generated by AF2 or RF or obtained from the PDB. The putative interacting region’s location on the protein surface is also known.
ChemNet was trained to reconstruct the proper atom positions from partially corrupted input structures when the learning challenge was framed as a structure-denoising task. This was accomplished assuming that all chemical knowledge about the modeled system was known from the beginning. The corruption strategy was tailored by researchers to the specific application of interest. The protein backbone coordinates, the amino acid sequence with side chain coordinates randomly initialized around the corresponding C-alpha atoms, and the small molecule chemical structure with atomic positions randomly initialized in the vicinity of the putative binding site are the inputs to the network in the case of the protein-small molecule docking.
By using an atom-centric method to anticipate the structures of small molecules and peptides in isolation as well in the context of a protein binding or active site described at the backbone level, it is possible to enhance protein binding and conformational heterogeneity. With the help of numerous sequence information to infer connections and a description of biomolecules, mostly at the residue level, networks like AF2, AF3, and RF can predict structures accurately. Nevertheless, the complexity increases significantly, and evolutionary information loses significance at the atomic level near the native structure.
The coordinates of the entire protein backbone surrounding a binding or catalytic pocket, together with an atomic-level description of the bound geometry of the interacting small molecules and amino acid side chains, would be a suitable place to start for an ensemble generation method. This network may be helpful for quickly evaluating and refining binding sites by simulating the conformations of peptides with constraints and small molecules in isolation and in a protein binding site.
After each iteration, ChemNet is trained using a mix of structure and confidence prediction losses. Every atom and every pair of atoms is considered when assessing the confidence of the modeled structures. The distance signed error approach is used to determine the accuracy of atom-atom pairs. Researchers anticipate all-atom lDDT scores as in AF2 on a per-atom basis. Furthermore, the network forecasts changes in the created model’s atomic locations 𝜎𝑖 (in Å) with respect to the reference structure. The expected RMSD can then be obtained by summing these anticipated deviations over a subset of atoms of interest, such as a small molecule.
Conclusion
ChemNet is a quick method that uses the protein sequence and backbone atom locations to create conformational ensembles of molecules. ChemNet does not predict the backbone structure of proteins, in contrast to other protein structure prediction techniques like AF3. This allows for faster calculations. This methodology encompasses not just biomolecules but also macrocycles and other intricate small molecules. For small molecule binder and computational enzyme design, the capacity to produce conformational ensembles for protein-small molecule assemblies is helpful. ChemNet’s evaluation of pre-organization and active site accuracy increases the discovery success rates for multi-step serine hydrolases. It is anticipated to be helpful in modeling complex non-protein molecules in protein contexts and isolation and in assessing protein-small molecule binder designs and enzyme models.
Article Source: Reference Paper | Code and neural network weights will be made publicly available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}