

Researchers from Jilin University have constructed a machine learning-based model that reduces confounders’ effects to identify potential driver genes involved in cancer initiation and progression.

Image Source: https://doi.org/10.1371/journal.pcbi.1010529
Cancer is one of the deadliest diseases worldwide. According to the World Health Organization (WHO), cancer has claimed the lives of nearly 10 million people in 2020. Mutations in the proto-oncogenes or the tumor suppressor genes can result in a cell exhibiting major cancer hallmarks. Therefore, identifying mutational genes plays a crucial role in comprehending various pathways involved in the conversion of a normal cell into a tumor.
Throughout the years, scientists have employed various experimental, sequence-based, and structure-based computational methods to identify cancer-driver genes. Different computational models such as ActiveDriver, DriverNet, TieDIE, and ResponseNet have been proposed and used to identify disease genes. However, these approaches ignore that not every correlation equals causation and that the external observed or unobserved confounding factors might also be involved in bringing about tumors.
A confounder is a factor that distorts the relationship between variables and the outcomes, thereby disrupting the results. For example, to understand the causal effect of a mutated gene in bringing about a cancer hallmark, such as uncontrolled cell proliferation, oxidative stress might be a confounding factor as it can affect both the mutation and cellular proliferation. In this case, it can be inaccurate to underscore the gene mutation as a true causal factor.
Therefore, to address this gap, the current study by Liu et al. has put forth a framework that estimates the Causal Effect of a mutation on cancer Biological Process (CEBP) to identify critical mutations by reducing the observed and unobserved effects of external (third-party) factors. Researchers have used the genomic and transcriptomic data from The Cancer Genome Atlas (TCGA) dataset to predict driver mutations that cause uncontrolled cell proliferation and epithelial-to-mesenchymal transition (EMT) that bring about metastasis across ten major cancer types.
Key Findings
The confounder-free CEBP model found major mutations in the genes involved in DNA replication and epithelial-to-mesenchymal transition as they drive cancer initiation and progression. The top ten genes with the highest mutation rates in each cancer type were selected and investigated for the causal role in cancer progression.
Throughout the study, mutations of the genes across ten major cancer types were investigated, including breast cancer, renal cancer, thyroid carcinoma, head and neck cancer, lung cancer, liver cancer, esophageal cancer, and bladder cancer.
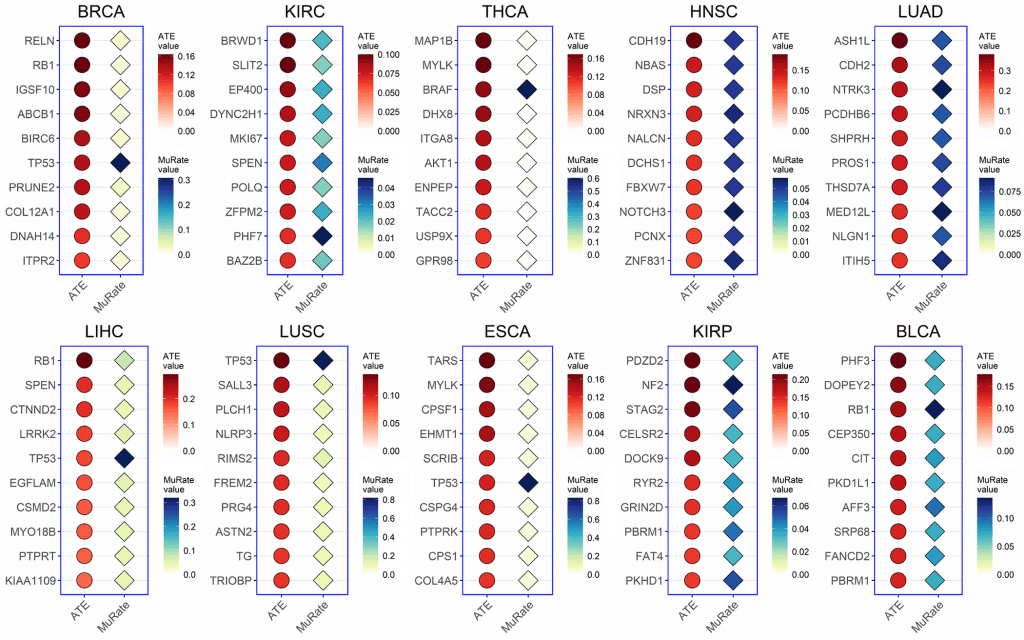
Image Source: https://doi.org/10.1371/journal.pcbi.1010529
The true causal effect of a mutation was determined by the Average Treatment Effect (ATE) score, where a positive score indicates that the mutation drives a biological process, and a negative score indicates that the mutation does not affect the given biological process.
Surprisingly, genes with the highest mutation rates did not correlate with a significant causal effect. For example, the high mutational frequency of VHL, a tumor suppressor gene involved in renal cancer, resulted in a very low causal ATE score, meaning the mutation is not entirely responsible for driving cancer initiation and progression.
Next, the researchers investigated genes with the highest causal ATE score, mutated and unmutated, across different cancers. It was found that the mutated genes with the highest score also played a pivotal role in cell proliferation and metastasis. Statistical analysis was also done on the same genes with the highest causal ATE score. It was established that these genes, either mutated or unmutated, did not affect their designated cellular processes.
For instance, the previously mentioned VHL gene, either in its mutated or unmutated form, is indeed involved in the replication of DNA and in the metastasis of renal cancer cells. This finding highlights that there is no significant relationship between mutational frequencies and their biological activity, leading to cancer.
Final Thoughts
The existing computational models ignoring the involvement of confounders bring a bias in the results, ultimately hampering the drug’s effect and causing poor treatment outcomes in cancer.
The current study bridges the gap and far exceeds the performance of other computational models using their causal effect-based approach. Newer potential genes found via this approach open up opportunities to further validate these genes’ role through wet experiments. This can also help in the development of more precise drugs that can hamper major cancer pathways, thus providing a better prognosis for cancer patients.
The authors of the study have also concluded with a promising way-forward note that a new de-conforming model which considers multiple mutations in the queried gene will be developed to mimic the complexity of the biological systems. This can help the scientific community better comprehend cellular mechanisms and malfunctions.
Article Source: Reference Paper | Codes and Data Availability: GitHub
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





{kind=link}