
Scientists can now model interactions and assess the possibility that proteins will form multimers or interact with other proteins or nucleic acids thanks to artificial intelligence (AI), which is transforming the prediction of macromolecular structures. As a result, there is an increasing demand for effective assessment tools. In this study, researchers from the Netherlands Cancer Institute introduced AlphaBridge, a suite of tools that predicts the surfaces of interactions between components of macromolecular complexes, giving scientists an easy way to examine and visualize relevant information. The predicted interfaces and intermolecular interactions, along with prediction confidence and sequence conservation scores, are summarized in these “AlphaBridge” diagrams. They are useful for quickly screening predictions, assessing the confidence of expected interactions, and ranking them before further in-depth (and resource-intensive) study.
Introduction
Algorithms for predicting protein structures have developed quickly, and they can now predict protein complexes as well. This revolution was led by AlphaFold, and AlphaFold2 first demonstrated previously unheard-of precision in predicting distinct protein structures. These advancements are now available to everyone because of the AlphaFold Protein Structure Database created by EMBL-EBI and Google DeepMind. At the same time, ColabFold provided all scientists with a quick and easy way to predict protein structures using an easy-to-use interface and powerful computing resources with effective search algorithms, matching AlphaFold’s accuracy on important benchmarks like CASP14 and ClusPro4. The ESM Metagenomic Atlas and RoseTTAFold were two notable independent attempts.
More about AI Models
A protein structure prediction program called AlphaFold-Multimer was created to simulate interactions between proteins. It was then expanded to protein complexes, allowing for virtual pull-down experiments and revealing novel biology in a limited group of proteins that are important in genome maintenance. About 2,700 ligands that are present in experimental structures were included in the AlphaFill approach’s database of possible complexes for predicted protein models. In order to anticipate protein complexes with nucleic acids and other biological ligands efficiently and dependably, AlphaFold3 recently introduced algorithmic advancements to the protein structure prediction problem. Despite restrictions on code access and licensing concerns, these recent AlphaFold advancements were integrated into an intuitive web server, making protein structure predictions more widely used. The frontiers of deep learning applications for the study of macromolecular complexes are being pushed by innovations such as NeuralPLexer, which directly predicts protein-ligand complex structures, and RoseTTAFoldNA, which studies protein-nucleic acid interactions.
Understanding AlphaBridge
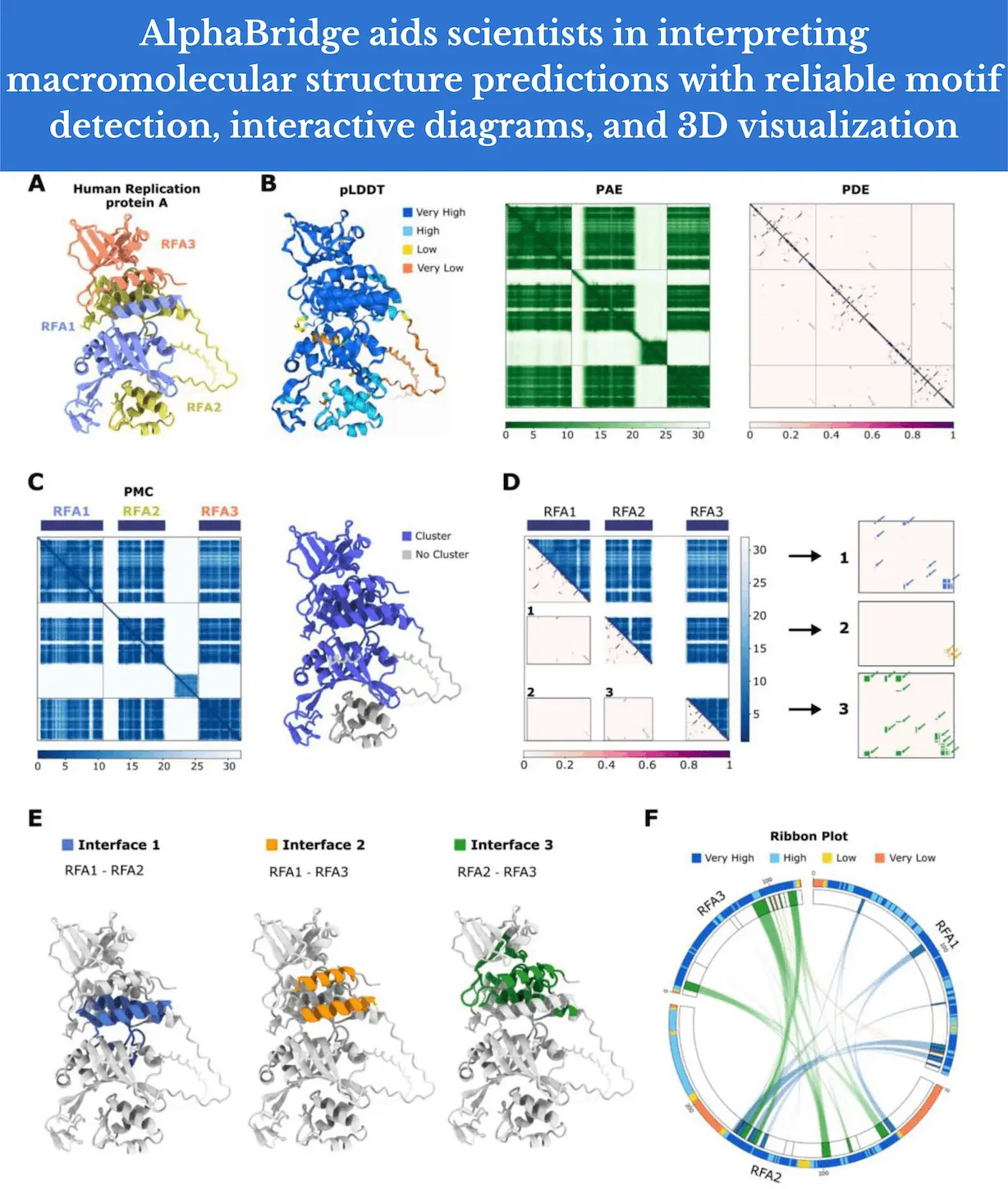
AlphaBridge is a set of tools developed by researchers to post-process and analyze data on interaction interfaces between predicted components of macromolecular complexes. The most pertinent data is then visually represented in a way that is easy for scientists who are interested in macromolecular complexes to understand. As of right now, AlphaBridge uses data from the AlphaFold3 server. Protein-nucleic acid (DNA and RNA) complexes are among the predictions it can process. An obvious extension is the processing of the different biological ligands that are available and the treatment of post-translational modifications (PTMs) in proteins. Both direct enhancement and integration of the method into other tools (such as OpenFold, ColabFold, or Predictomes) make it possible to process output from these servers.
A User-Friendly Approach: AlphaBridge
The probability contact matrix transcends the simple examination of the pLDDT and PAE matrix data by enabling the differentiation of trustworthy contacts in a reproducible and easily accessible way. The characterization of the linear motifs that comprise the contact linkages enables non-experts to easily examine the locations of interaction. The identification of these interactions with a single cut-off value as the threshold also shows promise for the development of new metrics that can score and rank the robustness of complicated predictions—a problem that is currently being worked on but requires more work.
As it has been used to illustrate interactions derived from cross-link proteomics applications, the circular layout visualization that the researchers used is familiar to many users and can potentially represent increasingly complex data in two dimensions. It can also be used to integrate additional information. The display of sequence conservation information (for all proteins) and AlphaMissense predictions (for human proteins), for instance, can be easily done. By adding these extra layers of information, the biological relevance of the interactions can be enhanced by analyzing and comprehending the functional consequences of interactions, identifying residues that are critical for a given function, or pointing to possible disease-associated variations.
Conclusion
AlphaBridge was created to help all scientists understand macromolecular complicated structure predictions. Taking into consideration the metrics that characterize the predictability of the predictions, it offers an automated, repeatable, and trustworthy technique to identify linear sequence motifs (contact links) engaged in binary interfaces between anticipated structures of macromolecules. On the other hand, it provides an interactive web interface that links live 3D visualization (fit for interactive analysis) with an easy-to-use method of presenting interactions in “AlphaBridge diagrams,” which help present information in presentations and publications. AlphaBridge aims to especially engage non-experts by making major advancements in the visualization and interpretation of AlphaFold predictions of macromolecular complexes.
Article Source: Reference Paper | The complete code is available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}