
Mads Jeppesen and Ingemar André from Lund University, Sweden, have demonstrated a protein complex assembly structure prediction pipeline conjugating Alphafold and symmetrical docking method. The approach makes structural apprehension of large multi-chain assembly of nature’s most complex symmetrical systems, the cubic symmetric group including tetrahedra, octahedra, and icosahedra, feasible at the atomic level. The proposed method can also be applied to cyclic, dihedral, and helical symmetry systems.
Exploring a Novel Methodology for Accurate Protein Assembly Structure Prediction
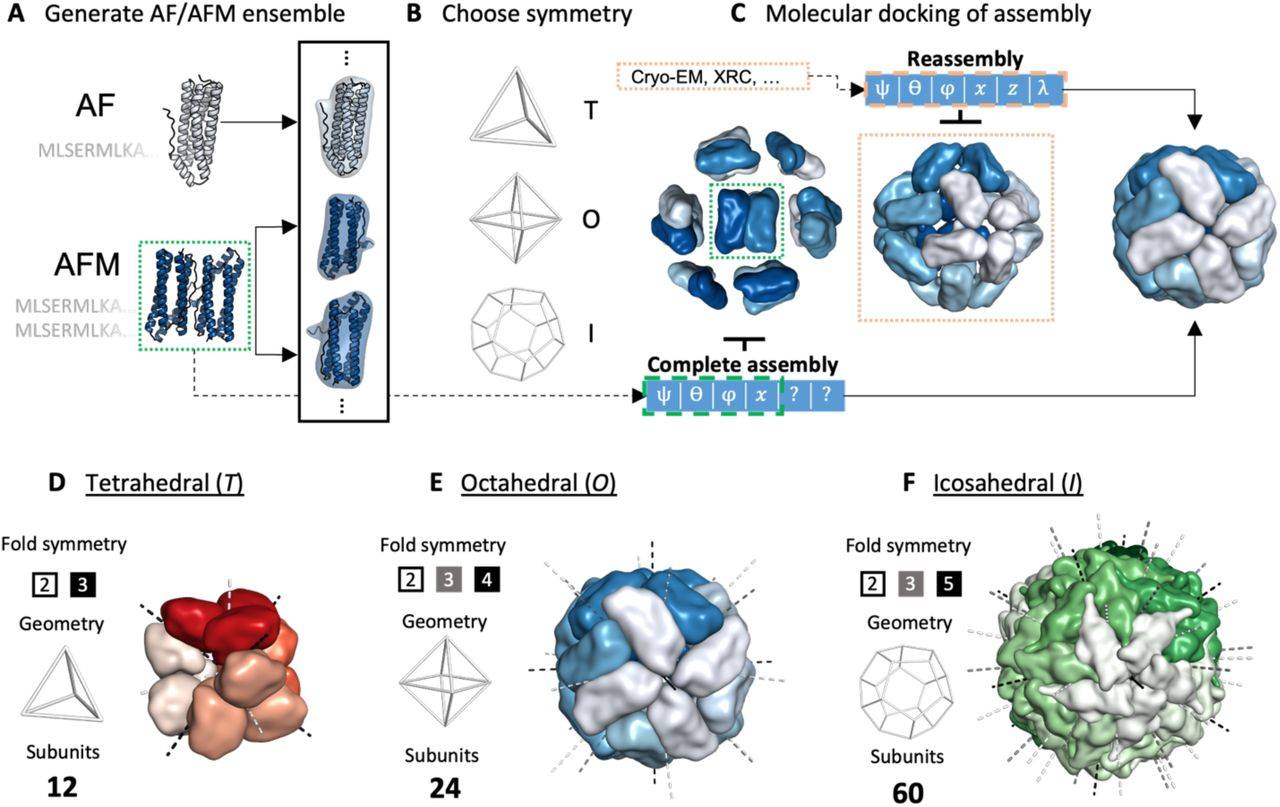
To gain a structural understanding of large protein complexes at the atomic level from their sequences, the authors carefully enunciate a novel methodology that displayed good TM (Template Modeling) Scores, was portrayed as high-quality according to DocQ scores, and had satisfactory RMSD (Root Mean Square Deviation) value and therefore, can be inferred as an accurate approach. EvoDOCK can generate energetically optimized models via the optimization of rigid body parameters of cubic systems when Alphafold2 yielded an ensemble of different subunits of a targeted protein complex from the sequences of the proteins of interest as input.
Two fundamental strategies for the model generating approach, reassembly and complete assembly, are established here, and for both cases, the mentioned pipeline yielded accurate outputs. Considering if the initial structural information or template of the proteins is unknown, the latter strategy should be utilized by creating a large parametric space sample directly from the amino acid sequence. If the information on protein structures is already present, then a local docking optimization is carried out around all rigid body parameters utilizing Rosetta Symmetric Machinery. In this scenario, if the protein structures are already there, predicting a native structural assembly is the most desired goal.
The research group of the same university, Daniel Varela, Vera Karlin, and Ingemar Andre, developed EvoDOCK in 2022, an efficient atomistic protein-protein docking algorithm for heterodimeric docking utilizing an evolutionary algorithm (EA) and Monte Carlo-based local search method. In this study, a symmetry version of EvoDOCK is used to build and refine complex homomeric symmetrical assemblies built from subcomponents predicted by AFM.
Structural Understanding of Protein Assembly: From Experimental Techniques to Computational Tools Delivering Unprecedented Accuracy
In physiological conditions, proteins often form complexes to execute cellular tasks, for example, DNA replication, transcription, translation, energy production so on; all require sophisticated assemblages and cooperation of multiple protein chains to fulfill vital roles. Understanding the structural details of the protein association is key to developing the notion about the mechanism and function of the complexes.
It is truly intriguing to capture the instances in the cellular machinery when and how proteins form large associations to execute their destined role, but the experimental approach wasn’t really unlocked until the onset of Cryogenic Electron Microscopy. The revolution in resolution has enabled scientists to obtain an enormous understanding of the microscopic biomolecular world. Solving protein structures from crystallographic techniques and NMR in the past decade unraveled many unknown aspects.
Despite the advancement in the instrumental world, experimental determination of protein structure is a laborious and time-consuming process, leave alone deciphering protein complex. But what if we can solve protein structure to the approximate accuracy of experimental settings from the desktop of our room without even stepping into a laboratory? In 2021, the launch of AlphaFold by the DeepMind team made this fantasy into reality.
The excellent collaboration of all the experimental efforts to explore the biological world and the advancement of Neural Network architectures made it possible. AlphaFold incorporates novel neural network architectures and utilizes training procedures based on protein structure’s evolutionary, physical, and geometric constraints. It elegantly solves the 3D reconstruction and the mirror image problems jointly by learning spatial transformations of the local reference frames of each of the protein residues.
Nevertheless, the scientific endeavor cannot halt here because exploring the scenario inside the cells is yet to be entangled fully. Certainly, AlphaFold2 is the breakthrough of this decade, but in the cells, proteins are dynamic entities, changing forms and spatial orientations very quickly to complete their function. AlphaFold2 can’t really accommodate this facet, perhaps due to the fact that experimental findings itself are limited in this aspect.
Moreover, regarding the associations of multiple proteins, although AlphaFold2 was not initially developed to model assemblies, surprisingly, several study groups reported positively about the ability of AlphaFold2 in those cases. Further, the DeepMind team modified it and developed AlphaFold Multimer (AFM) for this endeavor. But there is a constraint about amino acid chain length for AlphaFold Multimer due to limiting accuracy and memory capacity.
The researchers have pinpointed the major issue regarding the computation of protein complex models as the simultaneous requisite of predicting the interaction of multiple chains in a space and the overall organization of the chains, unlike the modeling of monomeric proteins. This drawback can be attributed to the fact that only a drop of protein assemblies is experimentally determined among the ocean of proteins in the proteome of organisms.
The Previous Endeavours of Determining Multimeric Protein Assembly
The authors mention an attractive strategy for building larger complexes was proposed by researchers from Science for Life Laboratory, Sweden, by sequentially assembling AFM-predicted dimeric and trimeric subcomponents by superposition. The issue that arises in this procedure is small errors while the prediction of individual interfaces of each subunit will propagate through the complexes and can result in severe clashes between subunits in the full assembly model. Also, modeling the complex having more than one unique interface region is challenging.
Some other groups published another novel approach of iteratively refining all interfaces of the assembly to minimize error propagation following searching for interactions not predicted by AFM using rigid molecular docking for dimeric associations. However, for large assemblies, this approach has not been explored before this study. This is likely due to the multitude of degrees of freedom involved in the search for optimal subunit placements, causing a computationally intractable optimization problem.
The researchers overcame the problem by making the combined AF/AFM docking approach tractable since multimeric assemblies display structural symmetry, which can therefore reduce the degrees of freedom substantially. Symmetry also becomes more prevalent as the protein complexes grow larger, accomplishing allostery, cooperativity, and multivalent binding.
Conclusion
AlphaFold2 has become a regular tool for computational biology-related studies, and combining AlphaFold2 with other complementary approaches has already facilitated innovative findings. In this regard, this study brings another dimension to understanding the wondrous function and association of proteins in diverse organisms by bringing the aspect of structural symmetry.
The researchers anticipate that the methodology presented here could be employed to study cubic assemblies in several different modeling scenarios. The energy landscapes of native assemblies could be investigated to understand the importance of subunit interfaces to the overall stability of the protein. Also, the method can be applied to predict cubic assemblies with unknown structures. The genetic material of viruses is stored inside a protein assembly known as the capsid protein. The pipeline can be helpful for investigating virus capsids as well.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.





{kind=link}