
Protein design has seen significant progress, but a comprehensive deep-learning framework for protein design, including de novo binder design and higher-order symmetric architectures, remains unexplored. Diffusion models have shown success in image and language generative modeling but have limited success in protein modeling due to the complexity of protein backbone geometry and sequence-structure relationships. The University of Washington researchers demonstrate this by optimizing the RoseTTAFold structure prediction network for tasks including protein structure denoising. In order to design therapeutic and metal-binding proteins, researchers have developed a generative model of protein backbones that performs exceptionally well in unconditional and topology-constrained protein monomer design, protein binder design, symmetric oligomer design, enzyme active site scaffolding, and symmetric motif scaffolding. Scientists experimentally characterize the structures and functionalities of hundreds of planned symmetric assemblies, metal-binding proteins, and protein binders to show the versatility and power of the RoseTTAFold diffusion (RFdiffusion) approach.
Understanding the Basics of Protein Design
The goal of protein design is to produce proteins with particular structural and functional characteristics, like catalytic sites, binding interactions, and topological folding. Robust machine learning models, known as denoising diffusion probabilistic models (DDPMs), have various characteristics that make them ideal for protein design. By denoising data tainted with Gaussian noise, DDPMs provide a variety of outputs that are quite similar to training data. With conditioning knowledge, they can direct the iterative generation process towards particular design goals. Rotationally equivariant DDPMs can model three-dimensional (3D) structures globally, regardless of frame, for protein design applications. Recently, by conditioning tiny protein motifs or secondary structure and block-adjacency information, DDPMs have been adopted for protein monomer design. Nevertheless, the low success of these initiatives in producing sequences that fold to the expected structures in silico is probably caused by the restricted ability of denoising networks to produce realistic protein backbones.
RoseTTAFold diffusion: A game-changing technique
Structure prediction techniques like AlphaFold2 and RoseTTAFold (RF) can be used to improve protein design by utilizing their profound knowledge of protein structure. Rigid-frame representation of residues, high precision, and an architecture that enables conditioning on design criteria at the individual residue, inter-residue distance, orientation, and three-dimensional coordinate levels make RF an excellent choice for protein design DDPM. RF is not useful for producing a restricted diversity of designs or simple site descriptions, nevertheless, due to its deterministic nature. It is possible to solve the issue of random noise and little initial structure information by fine-tuning RF as the denoising network in generative diffusion models. For this investigation, the denoising network design was based on an improved version of RF.
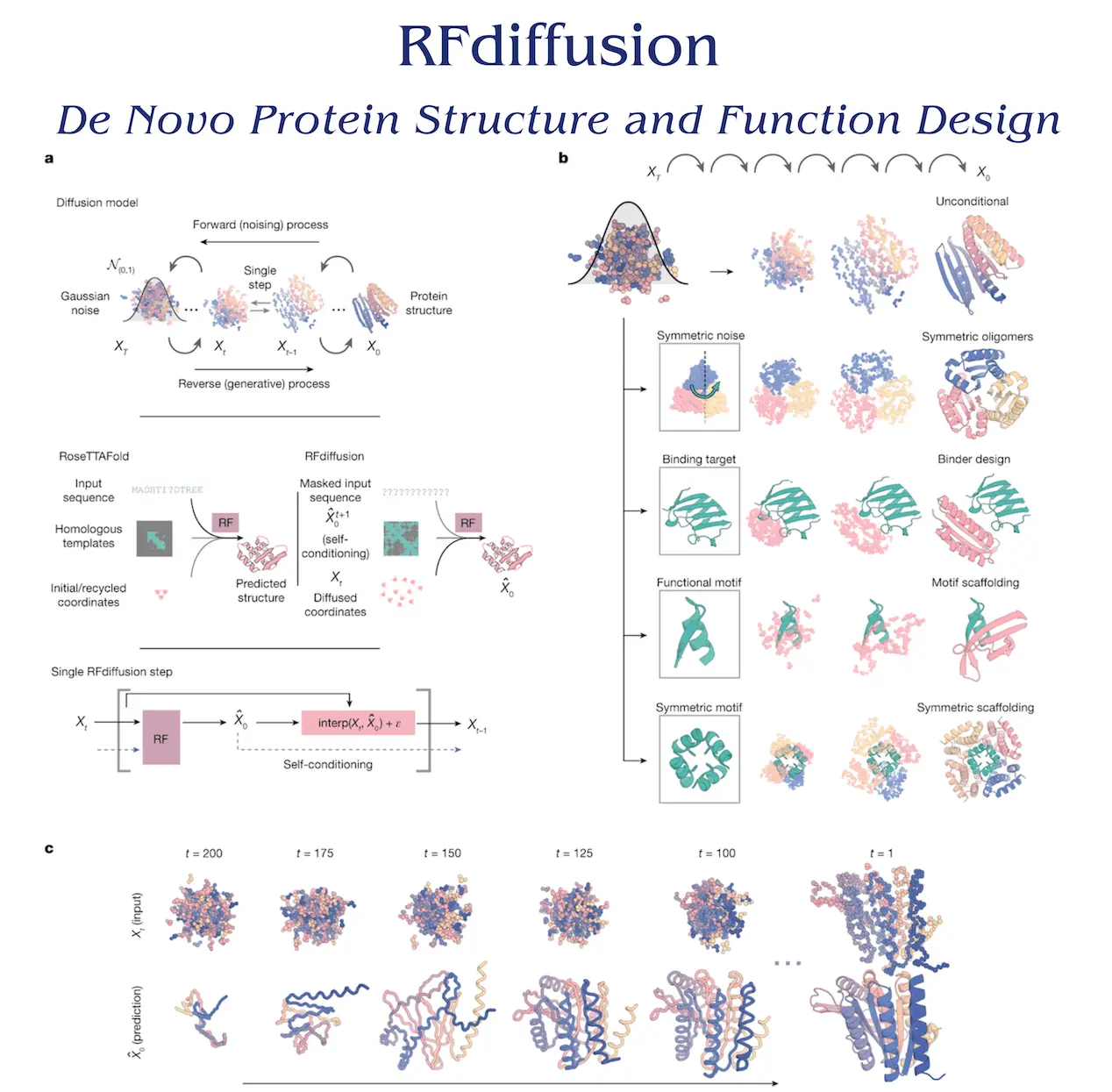
A Cα coordinate and N-Cα-C rigid orientation for every residue is utilized for training the RF-based diffusion model, RFdiffusion, using the Protein Data Bank (PDB). The training process of the model involves sampling noising structures for up to 200 steps, perturbing translations using 3D Gaussian noise, and utilizing Brownian motion to determine residue orientations. Denoising trajectories can match the data distribution at each timestep and converge on structures of designable protein backbones by minimizing a mean-squared error (m.s.e) loss between frame predictions and the genuine protein structure. In contrast to frame-aligned point error (FAPE), this method encourages the global coordinate frame to remain continuous across timesteps.
RFdiffusion is used to construct a denoised prediction, and random residue frames are initialized in order to create a new protein backbone. In order to provide input for the following stage, each residue frame is updated with noise. The amount of noise injected and the size of the reverse step are selected to correspond with the noising process’s distribution. RFdiffusion’s initial goal is to match all protein structures that are compatible with the completely random frames. However, after a few stages, the range of potential protein structures gets smaller, and RFdiffusion predictions start to look a lot like protein structures. Sequences encoding these structures are designed using the ProteinMPNN network, with an average of eight sequences sampled for each design. ProteinMPNN, in combination with the diffusion of structure alone, is taken into consideration, although it was not thoroughly investigated.
Networks convert coordinates into a predicted structure conditioned on model inputs in RF structure prediction and RFdiffusion denoising. Sequence is the main input in RF, with initial coordinates and templates providing additional structural information. Noised coordinates from the preceding stage serve as the main input for RFdiffusion, with additional conditioning information supplied for particular design requirements.
Two methodologies have been used to train RFdiffusion: self-conditioning and canonical diffusion models. The latter greatly enhanced performance on in silico benchmarks, motivated by the success of “recycling” in AF2. These performance gains could be explained by the enhanced coherence of predictions within self-conditioned trajectories. It proved more effective to fine-tune RFdiffusion using pre-trained RF weights than it was to train for the same amount of time using untrained weights. A successful in silico experiment is one that produces RFdiffusion output with high confidence worldwide within a 2Å backbone root-mean-squared deviation of the planned structure and on any scaffolded functional site within a 1Å backbone r.m.s.d. This is a stricter measure of in silico success than metrics based on template modeling scores that are employed elsewhere, and it corresponds with experimental success.
Applications of RoseTTAFold diffusion
- Unconditional synthesis of protein monomers – RFdiffusion may produce complex protein structures with no structural resemblance to training structures. There are several different designs, including mixed alpha-beta, beta, and alpha topologies. For de novo designs with 600 residues, the AF2 and ESMFold predictions agree well with the design structure models. For big proteins, RFdiffusion produces tenable structures, but because of its capacity for single sequence prediction, validation is challenging. RFdiffusion, which has been experimentally characterized, performs better than Hallucination with RF, a validated method that uses gradient descent or Monte Carlo search. With an NVIDIA RTX A4000 Graphical Processing Unit, RF diffusion production generates a 100-residue protein in 11 seconds, making it more computationally efficient than unconstrained Hallucination with RF.
Precision in crafting diverse topologies involved the creation of protein fold designs, including triose-phosphate isomerase (TIM) barrels, using the refined RFdiffusion method. For TIM barrels and NTF2 folds, the in silico success rates were 42.5 and 54.1%, respectively. Solvable, thermostable, and circular dichroism spectra that were in line with the design were seen during the experimental characterization of 11 TIM barrel designs.
- Synthesis of oligomers with higher order – The design of symmetric oligomers for use as catalysts, delivery systems, and vaccination platforms is of great interest. Structure prediction networks have been used to construct cyclic oligomers; however, because of lesser PDB representation, this strategy is ineffective for higher-order dihedral, tetrahedral, octahedral, and icosahedral symmetries. In order to overcome this, scientists develop symmetric oligomeric structures with any desired point group symmetry by generalizing RFdiffusion. Denoising predictions maintain symmetry due to the equivariant nature of RFdiffusion. RFdiffusion may produce symmetric oligomers with high in silico success rates even when it was not trained on symmetric inputs, especially when directed by an additional inter- and intrachain contact potential. Many of the RFdiffusion designs bear minimal resemblance to already solved protein structures, and they are almost identical to the AF2 predictions of the structures that the designed sequences have adopted.
At least 87 of the 608 experimental designs with SEC analysis had oligomerization states that were quite similar to the design models. The real structure is substantially similar to the design, as proven by negative stain electron microscopy (nsEM) data on a subset of these designs across multiple symmetry groups. RFdiffusion produced cyclic oligomers with alpha and/or beta-barrel structures resembling enlarged TIM barrels, offering a fascinating analogy between deep learning-driven innovation and natural evolution-driven innovation. One of the most prevalent folds in nature is the eight strands and eight helices that make up the TIM barrel fold. It is easier to investigate global variations in barrel curvature with RFdiffusion, which makes it possible to find TIM barrel-like structures with a lot more helices and strands. Structures with icosahedral, tetrahedral, and dihedral symmetry were also easily produced by RFdiffusion. According to SEC characterization, 38 D2, seven D3, and three D4 designs have the expected molecular weights. The overall topologies of the design models were consistent with the 2D class averages and 3D reconstructions of the D3 and D4 designs. Designs with strong assembly and several options for antigen display, like HE0902 (and future big assemblies of a similar nature), should be valuable as novel nanomaterials and vaccine scaffolds.
- Functional-motif scaffolding – RFdiffusion focuses on organizing protein structural motifs to provide the best possible function. During training and inference, motifs are entered as 3D coordinates, and scaffolds are constructed to store the motif atomic coordinates in place. RFdiffusion outperforms RFjoint Inpainting and Hallucination, solving 23 of the 25 benchmark challenges. RFdiffusion performs exceptionally well without the need for external potentials or hyperparameter adjustment, in contrast to Hallucination, which necessitates problem-specific optimization. When noise was not injected during the reverse diffusion trajectory, RFdiffusion-generated successful solutions with greater in silico success rates were found in 17 out of 23 issues. The inclusion of functional motifs in the RFdiffusion training set has no bearing on RFdiffusion’s capacity to scaffold them.
Conclusion
RFdiffusion is a significant advancement in protein design, outperforming most earlier techniques in terms of accuracy and complexity. It has a strong thermostability and produces a variety of unconditional patterns up to 600 residues in length, as anticipated by AF2. Unlike Hallucination approaches, which have been restricted to cyclic symmetries, RFdiffusion also permits higher-order designs with specified symmetry. When it comes to motif complexity, sidechain positioning accuracy, and motif recapitulation accuracy, RFdiffusion performs better than Hallucination techniques. Although it has been used to scaffold protein functional motifs using deep learning techniques, it is inaccurate and slow for big systems. High-throughput screening is laborious and costly; with atomic accuracy, functional proteins can be designed via RFdiffusion. By partial noising and denoising, it can also be expanded for protein model refining, allowing for tuneable sampling around a specified input structure. The capabilities and reach of RFdiffusion can be expanded to encompass ligands, nucleic acids, and particular design difficulties. With this method, de novo protein design can overcome the limits of natural evolution and reach higher complexity levels.
Article Source: Reference Paper | The code for executing RFdiffusion is accessible on GitHub, free for academic, personal, and commercial usage. Additionally, it is provided as a Google Colab notebook, with easy access through GitHub.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}