
DNA archives are a public repository for a great amount of knowledge about the evolution of bacteria and their mobile elements. However, the majority of this data is not well-constructed or is assembled inconsistently, which renders it inappropriate for extensive analysis. A consistently assembled set of 661,405 genomes was released by Blackwell et al. in 2021, and it was extended by 4.5 years until May 2023. The work also marks the start of an international cooperative endeavor to produce annotations for other species in response to requests from various research communities. Researchers from the European Bioinformatics Institute and European Molecular Biology Laboratory report in this work the first v0.1 data release of 1,932,812 assemblies (which is a combination of 1,271,428 new assemblies and the 661k dataset). For the 1.9 million genomes, the GTDB phylogeny has been consistently reprocessed for quality standards and taxonomic abundance estimations. An evolution-informed method is used to compress the entire set of genomes, bringing the size down to 102Gb in batched xz archives.
Introduction
The understanding of the planet’s bacterial organisms, which have developed over millions of years, is being drastically altered by the swift growth of sequencing technology in the bacterial domain. This technology emphasizes the significance of comprehending the bacterial domain in our world by offering insightful information about their biology, dynamics, and effects on the earth’s ecosystem. Bacterial genomes change via a variety of horizontal gene transfer mechanisms, such as viruses and mobile genetic elements like plasmids and transposons, as well as vertical inheritance, which occurs when parents split into pairs of offspring. This significantly affects how malleable and flexible they are. As little as 50% of the genomes of members of the same bacterial species can be shared; the remaining portion is accessory content. This optional additional material is made up of rare content that is transported by mobile elements and priceless cargo that offers essential adaptable characteristics. Research on the dynamics of functional elements and the variety of bacterial genomes is extremely valuable to the fields of clinical microbiology and public health.
The problem of inconsistent genome processing in the public domain has been addressed by the Environmental Network Analysis or ENA. This is caused by problems including batch effects, variable quality control (QC), and assembly using various tools and settings. Blackwell et al. set out to universally assemble, QC, and analyze all of the raw bacterial isolate whole genome sequence (WGS) data that was made accessible in the ENA in order to address this. The researchers involved research communities focused on particular genera/species in order to best serve the community as a whole.
Understanding AllTheBacteria
The AllTheBacteria project aims to update the 661k dataset and improve accessibility through a community-centric approach. A team of global researchers was involved in the promotion of the project on Slack and Twitter/X, as well as in the public microbiological bioinformatics channel. Initially, pipelines for open-source software were used to release assemblies, quality control, taxonomy data, and search indexes. With plans for the next releases, the project has merged communities. Reduced accessibility was achieved by compressing the 661k assemblies to 20Gb and the indexes to 100Gb using the phylogenetic compression approach, which was also employed to minimize data storage.
Methodology
The study used Illumina bacterial isolate whole genome sequence raw sequence datasets from the ENA as of May 2023. Reads were downloaded using either prefetch/fasterq-dump from the SRA-toolkit or enaDataGet. The genome assembly pipeline was the same as that used by Blackwell1, a wrapper around Shovill. Taxonomic analysis on isolate data is simpler than on full metagenomic data, and the major species, its relative abundance, and the nature of contaminants are established. Simulation experiments with mixtures of different species at different abundances were conducted, and it was determined that sylph was more accurate, faster, and required less RAM than the tools used for the 661k (Kraken/Bracken) (Sylph version 0.5.1).
A species call was made using a lookup table created using TaxonKit and GTDB taxonomy data from the “Genome file” column of the sylph output. Sylph produced no output for the readings from 3,252 samples, most likely due to the lack of matches to the reference database. Every assembly was subjected to a run of CheckM2 version 1.0.1 and the program assembly-stats, which collected basic data for each assembly.
MiniPhy, which enhances compression through intelligent batching of genomes, was used to compress all assembly FASTA files. The procedure entails splitting the genomes into batches of roughly equal size, usually according to species. Each batch was then subjected to a run of MiniPhy, which produced an estimated phylogenetic tree and rearranged the genomes to improve compression. The high-quality assemblies were sketched at k=14 using pp-sketchlib v2.1.1, which allows sequence similarity search through computing a Jaccard index.
Result of the Study
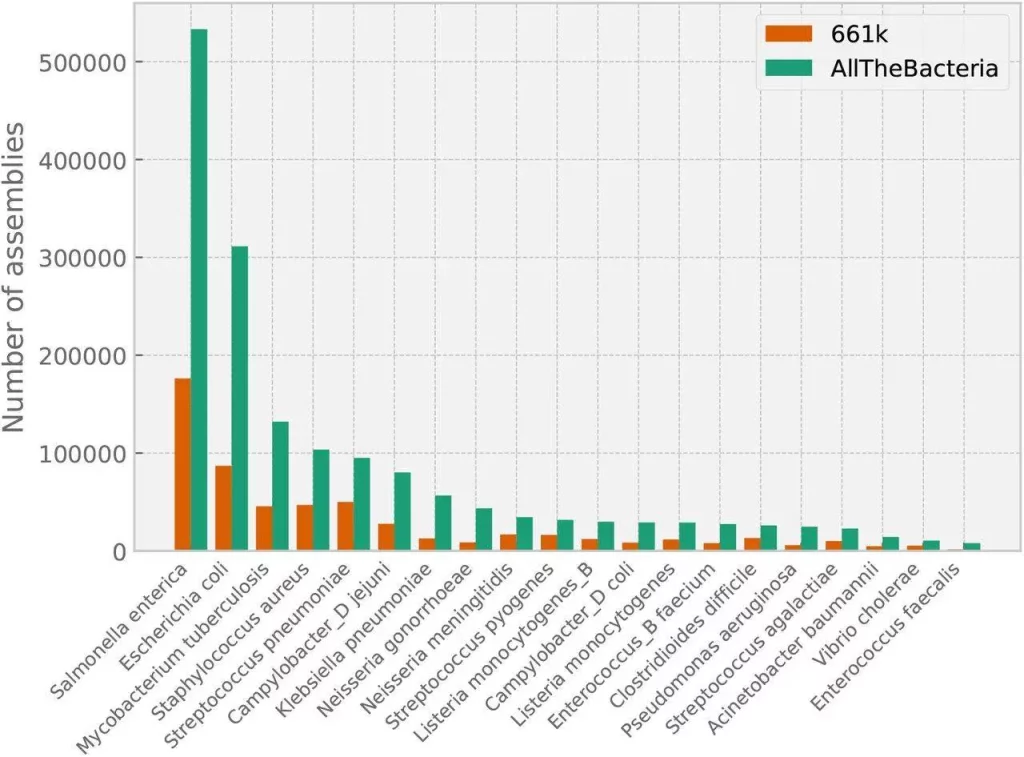
When the 1,271,428 additional assemblies produced by the project are added to the 661k dataset, a total of 1,932,812 assemblies are obtained. With the transfer of the NCBI taxonomy from the 661k project to GTDB, all sample sequence reads were reprocessed. The requisites, which include a genome size between 100k and 15Mb, no more than 2000 contigs, N50 at least 5000, majority species at above 99% abundance, CheckM2-completeness at least 90%, and CheckM2-contamination of no more than 5%, are met by a file list of 1,857,792 high-quality assemblies. Seventy-seven percent of the high-quality dataset is made up of the top 10 species of significant clinical interest, which dominate the data.
Users were able to compress assemblies effectively without the need to install additional software by using batching and special software. Inexperienced users used 3 Terabytes of disc space for each assembly. This was reduced to about 380Gb by batching by species into groups of 4000 genomes and using xz as a compressor. Disc utilization was lowered to 102Gb by using the MiniPhy tool for more efficient batching before compression.
Conclusion
A collaborative project titled AllTheBacteria has made excellent data and assemblies 0.1 available. Subsequently, work packages pertaining to gene annotation, pangenome creation, mobile element and phage analysis, and gene/feature identification will be sent to various groups. Additionally, the project offers indexes like COBS, sourmash, and the snakemake workflow mof-search, which combines minimap for full alignment against the full data with compressed COBS indexes. The aim is to ensure the assemblies and first search index (ppsketchlib) are immediately useful.
Article source: Reference Paper | Data Availability: All assemblies and metadata from Release 0.1 | Assembly Pipeline
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}