
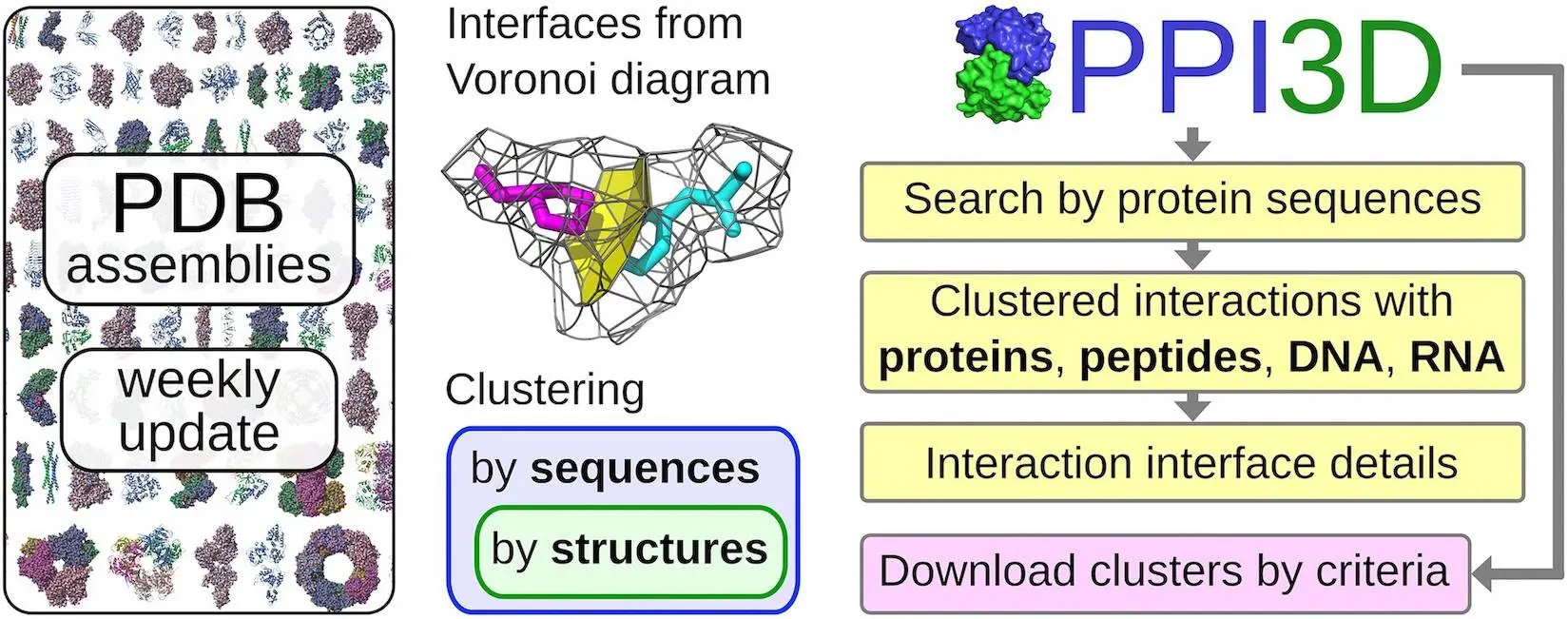
Understanding molecular mechanisms requires an understanding of protein interactions with nucleic acids. Researchers can query preprocessed and clustered structural data, analyze the data, and draw conclusions about protein interactions based on homology using the PPI3D web server. All interactions for proteins homologous to the query, interactions between two proteins or their homologs, and interactions within a particular PDB entry are the three interaction exploration options that the server provides. Protein annotations, structures, interface residues, and contact surface regions are all included in the interactive study. All contact interfaces and binding sites from PDB are included in the weekly updated PPI3D database, which is grouped according to structural similarity and protein sequence. For proteins that are extensively investigated, this prevents repetitive information by producing non-redundant datasets without sacrificing different interaction types.
Introduction
Proteins are essential for many biological activities; they frequently form complexes with ligands, peptides, nucleic acids, and other proteins. For a thorough comprehension of biological processes, one must comprehend these interconnections. Protein complexes can have their structures determined experimentally or predicted computationally, and both methods are available in the Protein Data Bank (PDB). Millions of individual protein structures have been correctly predicted thanks to recent advances, but protein-protein and protein-peptide complex structure prediction is still difficult. For experimentalists and computational biologists creating protein complex modeling techniques, using experimentally determined structures is essential.
A valuable resource for structure-resolved structures is the PDB, which offers a wealth of interaction data for protein complexes. Redundancy and the requirement to separate biologically meaningful interactions from crystal packing might make it difficult to obtain and analyze this data. It can be challenging to obtain representative monomeric structures when a protein complex has many PDB entries, leading to various instances. Because of the interface definition and similarity metric, structure-based comparison is necessary to generate representatives for interaction interfaces, and it is not easy to reduce the redundancy of interaction data by simple sequence-based filtering.
Many web-based programs have used PDB interaction data to comprehend the connections and functions of proteins. Biologically significant protein-protein interfaces and assemblies can be found in crystal structures using programs such as PISA, EPPIC, ProtCID, and ProtCAD. Nevertheless, there is no infallible technique to prevent the incorrect assignment of biologically significant assemblies or interfaces. PDB interaction data is annotated and categorized by other tools, which then use the data to infer novel interactions or transfer 3D information to homologs. A few examples are DNAproDB, which specializes in the categorization and annotation of protein-DNA complexes; HOMCOS, which concentrates on searching and template-based modeling of homologous protein complexes; and 3did, which connects PDB interaction data to Pfam domains. Protein-protein interaction networks are enhanced with 3D structures by web servers such as Interactome3D, LEVELNET, and Proteo3Dnet.
Understanding PPI3D
A non-redundant set of pairwise interactions created from a current set of PDB biological assemblies is provided by the updated PPI3D service, enabling users to thoroughly search through and analyze the results. Additionally, it enables users to build template-based models and draw conclusions about the interaction sites of the query proteins based on homology. In order to define the contribution of each residue-residue contact to the interaction interface, PPI3D derives and analyses interaction interfaces using Voronoi tessellation. Additionally, by discovering alternative interactions that would be missed in clustering based solely on sequence similarity, the method allows robust structure-based clustering of interfaces and binding sites. There have been two main enhancements: the ability to customize sets of interaction interfaces and the expansion of capabilities to protein-nucleic acid interactions.
Key Features of PPI3D
In recent CASP and CAPRI research, the server PPI3D achieved excellent results in protein assembly modeling and has been a key component of both computational and experimental studies. Its capacity to investigate the larger structural background and test theories on particular residues implicated in binding is still helpful. Since accurate structure prediction of protein-nucleic acid complexes is still mostly refractory, PPI3D’s capacity to deliver homology-based inferences linked to protein-DNA or protein-RNA interactions is particularly relevant.
- PPI3D is a unique set of tools that analyzes structure-resolved data on biomolecular interactions.
- It uses precomputed, non-redundant structural data updated weekly to keep in sync with PDB data, unlike most other servers that rely on PDB data that are several months or years old. PPI3D offers sequence-based searches that can detect close or distant interacting homologs.
- The user interface allows interactive analysis of diverse interactions for proteins of interest at both sequence and structure levels.
- The analysis ranges from general data on identified interfaces/binding sites to properties of individual residue-residue contacts.
- PPI3D is the only server that uses a rigorous Voronoi tessellation-based methodology for clustering and analysis of interactions.
- The new option to download all data on interaction interfaces is useful for large-scale analyses and for training and testing machine learning methods for predicting structures or properties of macromolecular complexes.
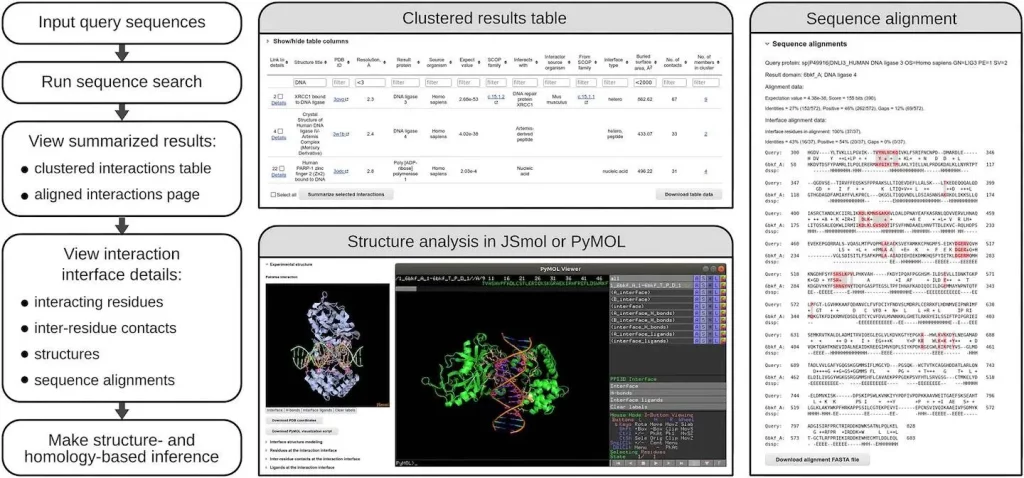
Image Source: https://doi.org/10.1093/nar/gkae278
Analyzing Protein Interactions
The vast diversity of protein-protein interactions presents a multitude of binding modes, which can be missed by databases that only cluster interactions by protein sequences. PPI3D, a database that clusters interaction interfaces by sequence and structure, enables the identification of these alternative binding modes. One such mode is the dual binding protein pairs, cohesion, and dockerin, found in a cellulosome complex. The biological significance of this mode is still unknown, but it can be regulated by pH. A PSI-BLAST search in PPI3D revealed 32 structures that can be clustered differently. A more stringent clustering produced 17 clusters, while a lenient clustering produced only 12 clusters. Changes in protein binding modes can occur upon ligand binding, such as agonist binding, which causes large conformational changes in the human GABAB receptor. PPI3D clustering recognized these two interfaces as distinct clusters.
Finding DNA Binding Nodes by PPI3D
Enzymes known as DNA polymerases are vital for the synthesis of DNA during replication and repair. The most common B-family DNA polymerases function as DNA polymerases and as three to five exonucleases in various domains. PSI-BLAST was utilized by researchers to look into the interactions these enzymes have with DNA. They discovered 271 protein-nucleic acid binding sites in polymerases from bacteria, viruses, eukaryotes, and archaea. Polymerases attach DNA in two separate modes, which correlate to DNA synthesis and proofreading, as stated in the study.
Conclusion
A user-friendly environment for searching and analyzing structure-resolved protein-centered interactions is provided by the PPI3D online service. If there is no interaction data available for the protein(s) of interest, PPI3D might be extremely useful. A single-sequence mode of sequence searches can aid in the inference of potential interaction partners by identifying homologs coupled to other proteins, peptides, or nucleic acids based on their identified structures. Similarly, the interactions found in the “two sequences” search mode can indicate a possible interaction between two query proteins. In both situations, template-based models and sequence alignments with the identified structural homologs can be used to investigate these preliminary theories further at the residue level.
Article Source: Reference Paper | PPI3D is available on the web server.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}