
Information about the function and three-dimensional structures of proteins can be found in the AlphaFold Protein Structure Database (AFDB). Almost all of the proteins in UniProt have full-length predictions provided by it, demonstrating the intricate connections between domains and folding units. Characterizing 200 million structures is still a struggle, despite AFDB’s potential. Here, researchers introduce The Encyclopedia of Domains, or TED, which divides and categorizes domains throughout the AFDB by combining cutting-edge deep learning-based domain parsing and structure comparison techniques. Over 3656 million domains are described by TED; these are more than detectable using sequence-based techniques. The set of known protein structural domains increases because over 80% of TED domains are identical to recognized superfamilies in CATH. In addition to expanding domain coverage to over 1 million taxa and revealing hundreds of folds and architectures throughout the protein fold space, TED also reveals 10,000 invisible structural connections.
Introduction
The protein structure universe has grown to over 200 million sequences thanks to AFDB. This UniProt-based protein structure database has improved both commercial and academic research in the life sciences. For next-generation structure-based drug development that uses cutting-edge technology and unique data, the database’s 3D depiction of protein structures is essential. Studies on AFDB were carried out by Bordin and Schaeffer, who divided full-length models into related proteins and described their roles. While categorizing domains under the CATH and ECOD frameworks, they also looked at the makeup of particular proteomes, concentrating on the original AFDB model organism dataset.
Gene3D and the Pfam database are two structure-based methods for domain discovery that concentrate on protein families represented by Hidden Markov Model (HMM) profiles and multiple sequence alignment (MSA). Although sequence-based discovery provides more coverage, it is frequently ineffective on distant cousins because of its limitations in HMM detection capabilities. Although low experimental structures have restricted structure-based assignments, they can show distant relatives and allow for higher-quality domain boundaries.
Researchers in this work thoroughly examine the domain composition for the whole AFDB (version 4). This description includes more than 371 million putative domains, derived from over 214 million protein sequences in over 1 million taxa. Three automated parsing techniques—Merizo, Chainsaw, and UniDoc—agree to identify these domain structures. Furthermore, over 251 million domains can be positioned on the CATH hierarchy by using structural comparison techniques like Foldseek and an internal deep learning method called Merizo-search.
Looking into TED
More than 1 million distinct taxa with over 214 million UniProt sequences and protein lengths up to 2700 residues have predicted structures in the AFDB V4. Researchers did an analysis procedure by eliminating the structures of similar sequences, which resulted in a collection of 188 million non-redundant sequences from 653,460 species that scientists call TED-100.
Three state-of-the-art domain parsing techniques and structure classification algorithms are combined in the workflow to find known domain folds in the AFDB. More than 100 million ‘TED’ domains were found using this method than using sequence-based techniques, totaling 371 million. According to TED-100, single and multidomain proteins are separated 42:55, with the latter having up to 20 domains. While 26.2% of Pfam targets and 33.9% of Gene3D targets lack recognizable domains, only 2.8% of TED-100 targets do so. The percentage of non-domain residues (NDRs) varies by superkingdom; in bacteria and archaea, it is about 10%, while in eukaryotes, it can reach 30%.
Only over 2% of domains fall into the lowest bin, according to an analysis of the average plDDT scores for the TED-100. Most domains, however, fall into the “very high” to “high” bins. The domain identification workflow appears to be successful in identifying plausible domains within the well-folded portions of AF2 models, as the domain segmentation algorithms do not take residue plDDT into account when determining domains.
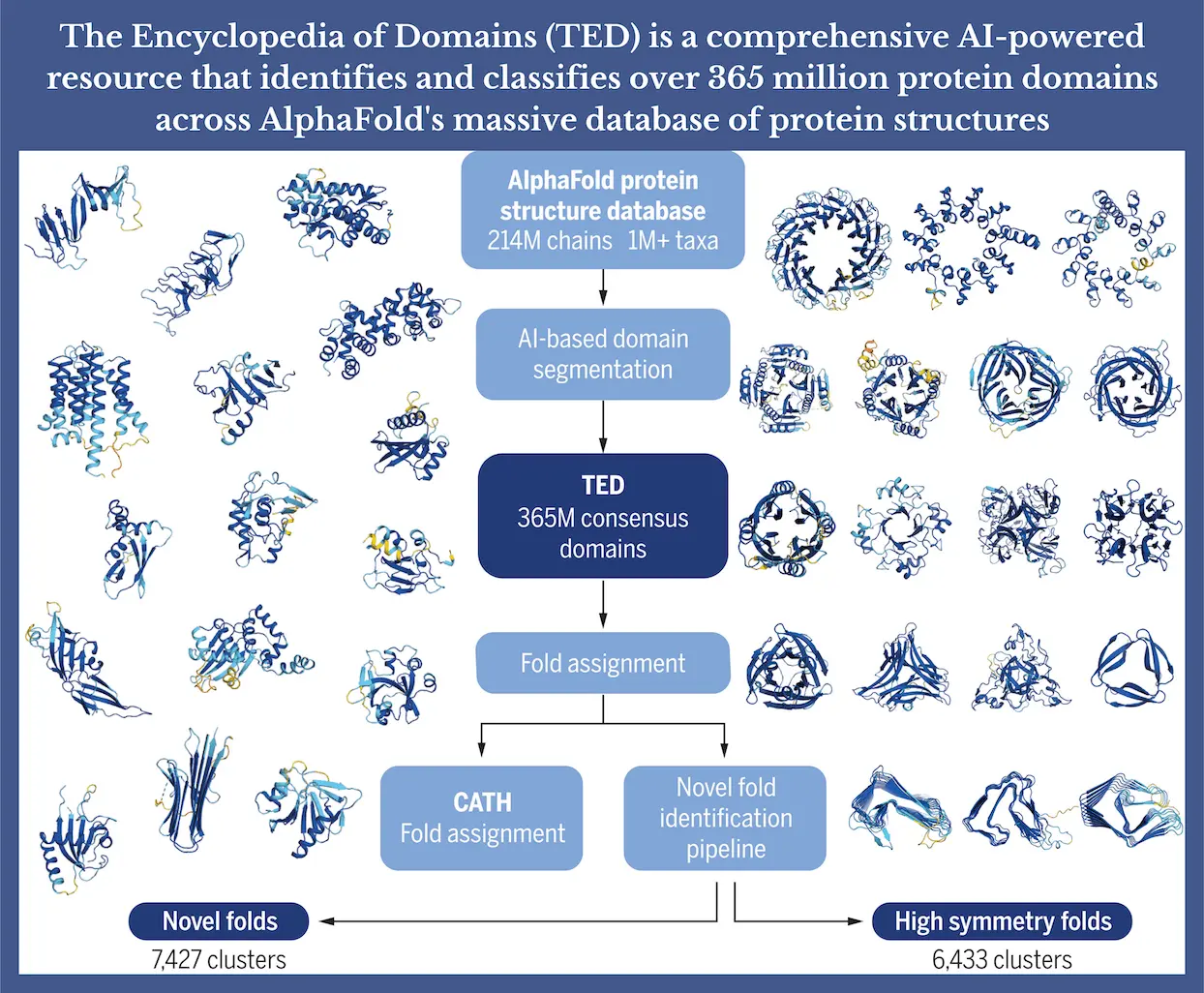
Workflow of TED
The AFDB database has 214 million target sequences, which are filtered to identify 188 million non-redundant targets (TED-100) and a set of redundant targets (TED-redundant). These domains undergo automated domain parsing, with assignments derived from consensus among three methods. TED-100 domains are processed by MMseqs2, creating over 121 million clusters at 50% identity. Domains are matched to CATH domains via Foldseek and Merizo-search, and categorized into superfamily, topology, or no-matches. Clusters are annotated with CATH labels, creating partially labeled and unlabelled clusters. Low-quality domains in unlabeled clusters are filtered out. Resultant domains undergo a new workflow for identification, involving clustering and database searches for matches to known structures. Poor-quality domains are identified using an in-house deep learning method, while novel domains are scored on internal symmetry using the SymD program. Full-length targets undergo automated domain parsing by Merizo, Chainsaw, and UniDoc, with a consensus taken by identifying assignments where three (high), and two (medium) methods agree or no consensus is found (low). The “TED” count combines TED-100 and TED-redundant, including 13 million overlapping targets.
Conclusion
The enormous volume of data researchers had to handle in the AFDB served as the primary driving force behind several algorithmic decisions made by the researchers. As a result, scientists want TED to be a continuous evolution that changes in response to data and user demands. The objective is to give the community the most thorough explanation and dissection of the AFDB’s structures. Researchers anticipate that TED will serve as a foundation for a wide range of investigations, including offering a sizable dataset for the training and testing of a new wave of structural biology applications based on deep learning.
Article Source: Reference Paper Abstract | Full Preprint | The code for the TED website is available on GitHub.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











 segments and classifies 370M protein domains from AFDB, revealing new structural insights.){kind=link}