

The algorithms that track users' Netflix viewing habits could soon be used to track cancer development in order to direct doctors in managing cancer.
An international team of researchers led by Dr. Nischalan Pillay (UCL Cancer Institute) and Dr. Ludmil Alexandrov (University of California, San Diego) used AI to identify 21 frequent faults in the structure, order, and quantity of copies of DNA present when cancer begins and progresses. These widespread errors, known as copy number signatures, could help doctors find medicines that match the tumor’s characteristics.
As the Netflix algorithm suggests new videos on the basis of a person’s like and dislikes, the researchers developed a similar algorithm that can filter through thousands of lines of genomic data to find common patterns in the way chromosomes organize and arrange themselves. The system may then classify the patterns that develop, assisting scientists in determining the types of cancer faults that can form.
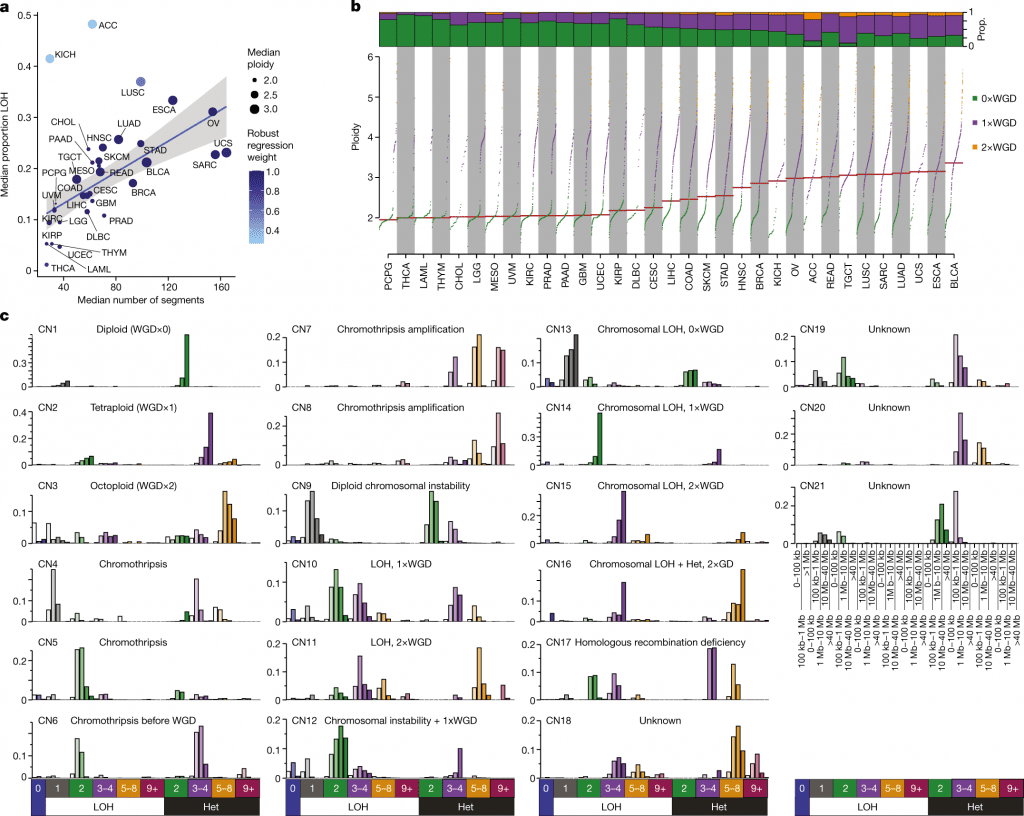
Image Source: Signatures of copy number alterations in human cancer
DNA alterations, such as gains and losses, frequently occur in cancer and result from a variety of interconnected events, including replication stress, mitotic mistakes, spindle multipolarity, and breakage–fusion–bridge cycles, which can cause chromosomal instability and aneuploidy.
These copy number changes are involved in developing, spreading, and evolving therapeutic resistance in cancer.
In the given study, the scientists present a conceptual framework that can be broadly applied to a variety of data types, including whole-genome sequencing, whole-exome sequencing, reduced representation bisulfite sequencing, single-cell DNA sequencing, and SNP6 microarray data, to analyze the patterns of copy number alterations in human cancer.
By applying this paradigm to 9,873 tumors from The Cancer Genome Atlas, which represent 33 different forms of human cancer, it was possible to identify a set of 21 copy number signatures that account for 97 percent of samples’ copy number patterns.
The biological processes of whole-genome duplication, aneuploidy, loss of heterozygosity, homologous recombination deficiency, chromothripsis, and haploidization were each associated with one of seventeen copy number signatures.
Four copy number signatures’ causes are still unknown. Amplification signatures for extrachromosomal DNA, disease-specific survival, and proto-oncogene gains like MDM2 are seen in some cancer types.
No copy-number signature was connected to numerous well-known external cancer risk variables, in contrast to base-scale mutational signatures.
By exposing a variety of mutational pathways that result in these modifications, the researchers’ findings synthesize the worldwide landscape of copy number alterations in human cancer.
Development of Highly Complex Cancer Genomes
Beyond changes to a single chromosome, whole-genome doubling (WGD) and chromothripsis can also change the genomic copy number.
While chromothripsis is a “genomic catastrophe” that results in clustered rearrangements connected to oscillating copy number patterns, WGD is the process by which a cell’s complete chromosomal material is duplicated from a diploid to a tetraploid state.
During the growth of tumors, these evolutionary processes may repeat at various intensities, producing very complex cancer genomes.
The Development of a Previous Computational Framework
To separate somatic mutations into the mutational signatures of single base substitutions (SBSs), doublet base substitutions (DBSs), and minor insertions or deletions (IDs), the scientists previously built a computational framework.
Unprecedented insights into the external and endogenous processes that shape cancer genomes at the single nucleotide level have been gained by analysis of mutational signatures.
Prior research has also looked at the hallmarks of genomic rearrangements in cancer, and these investigations have provided insights into cancer-subtype-specific HRD and templated insertions.
Additionally, improvements in signal-to-noise ratios for identifying cancer processes have been made due to the bioinformatics integration of single nucleotide mutations, rearrangements, and microsatellite instability profiles.
However, the translational applicability of rearrangement signatures is severely constrained because they can only be obtained from whole-genome sequencing (WGS) data.
The Development of a Mechanism-Agnostic Approach
Since a priori knowledge of the mutational processes active in those tumors remained unknown, the scientists recently established a “mechanism-agnostic” approach to summarise allele-specific copy number patterns in whole-genome sequenced sarcomas.
Breast cancer, ovarian cancer, prostate cancer, multiple myeloma, and other cancer subtype-specific techniques have been used to examine copy number patterns using known hallmarks of genomic instability.
The researchers were aware of no method that enabled the probing of copy number signatures produced from allele-specific profiles across various cancer types and experimental assays at this time.
To fill this gap, they created a new framework to interpret copy number signatures across several experimental platforms and cancer types.
The Endpoint
In this study, the researchers introduced a framework for copy number signature analysis that is superior to mutational signatures of substitutions, IDs, or rearrangements in its ability to explore copy number trends across a variety of cancer types and experimental platforms.
In general, signatures of substitutions and IDs may be extracted from WGS, and at a considerably lower resolution, WES data have been detected across the majority of cancer types.
Rearrangement signatures cannot include crucial prognostic information like WGD and can only be obtained from WGS data.
The robust and reliable detection of copy number signatures using WGS, WES, reduced representation bisulfite sequencing (RRBS), SCS, and SNP6 microarray data was made possible by the copy number signature framework, which may be used across all cancer types.
For patients in whom amplicon signatures are seen, the detected copy number signatures have clinical significance and prognostic consequences.
Furthermore, the discovery of a copy number signature linked to HRD—although not the first such discovery—indicates that the accuracy of these tests may be further improved by adding such signatures to the current bioinformatics tools for predicting HRD.
The field of copy number signatures is young; several unique methods have already been used in various tumor types. Which models are more effective at answering particular clinical or biological problems will become more apparent as the field develops.
Pan-cancer analyses incorporating all of these techniques will be crucial to answering these concerns, and the scientists presented in this study the first step in that direction: a collection of copy number signatures created from allele-specific profiles that are independent of mechanisms.
Article Sources: Steele, C.D., Abbasi, A., Islam, S.M.A. et al. Signatures of copy number alterations in human cancer. Nature (2022).
DOI: https://doi.org/10.1038/s41586-022-04738-6
https://www.ucl.ac.uk/news/2022/jun/netflix-style-algorithm-builds-blueprint-cancer-genomes
Learn More About Bioinformatics:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}