
Targeted and efficient treatments for identified protein targets necessitate the use of structure-based drug design (SBDD); this is still a challenge owing to the vast chemical space and complex protein-ligand interactions. The progress of language models (LMs) in NLP is commendable; still, there is much to learn about how LMs can be utilized in SBDD. To help narrow this gap, scientists at Texas A&M University present Frag2Seq, which uses the LMs for SBDD and builds molecules based on fragments mapping to functional modules. The immediate input comprises 3D molecules, which are transformed into fragment-informed sequences using the fragment local frames and SE(3)-equivariant molecules; the model also reclaims geometric information from 3D fragments. Target-aware molecule synthesis is enabled by the integration of reversed sequences involving protein pocket embedding from an inverse folding model. The model that does exceedingly well on chemical characteristics, binding vina score, and selectivity is the one that generates drug-like ligands with enhanced binding to target proteins. Besides, it has a higher sample efficiency compared to baselines that use diffusion and autoregressive atoms.
Introduction
Machine learning, particularly in the recent couple of years, has been observed to have the potential to advance scientific research. Machine learning methods are unstable in SBDD because the chosen model should predict protein-ligand interactions and optimize the created compounds’ properties to be drug-like. Recurrence structures have previously been used in diffusion models as well as autoregressive models in an attempt to create molecules and encode context information. However, diffusion models often take thousands of steps to generate, which means that they generate inefficiently, and these diffusion models only define generation at the atom level. Producing molecules is another area of work that starts with molecular fragments; choosing the fragments and joining them requires quite complex neural networks, which are often multiple, forming a pipeline.
About Language Models
LMs, with their data processing and generative ability, have promising potential in many domains. Large language models, as researchers know, are very good at understanding complex patterns, making them very suitable for complex natural language processing tasks. However, there are a number of things about incorporating LMs with SBDD that are still not very clear. LMs have some advantages, such as the generation of highly specified drug-like molecules, learning from vast biological and chemical articles, and quick handling of enormous data sets. Some of the issues are duplicated physical and chemical representation of molecules, fragment-based synthesis, including contextual protein information into the LMs, and converting LMs to accommodate the geometric graph topology of the molecular data. By tackling these problems, SBDD may be able to realize the full potential of LMs, which should transform the process of drug discovery and development into one with a higher rate of success.
What is Structure-based Drug Design (SBDD)?
Structure-based drug design (SBDD) is one of the most important strategies employed in medicinal chemistry. It involves designing and optimizing small molecules to engage with biological targets, commonly protein binding sites, in a rational and efficient process. It involves online interaction to develop new therapeutic medications as it involves designer interactions of systems biology that may reduce the detrimental consequences and optimize effectiveness. Virtual screening and the subsequent experimental compliance have been constitutive of SBDD from the beginning. Nevertheless, such procedures may require a lot of labor and time and may be relatively expensive, too.
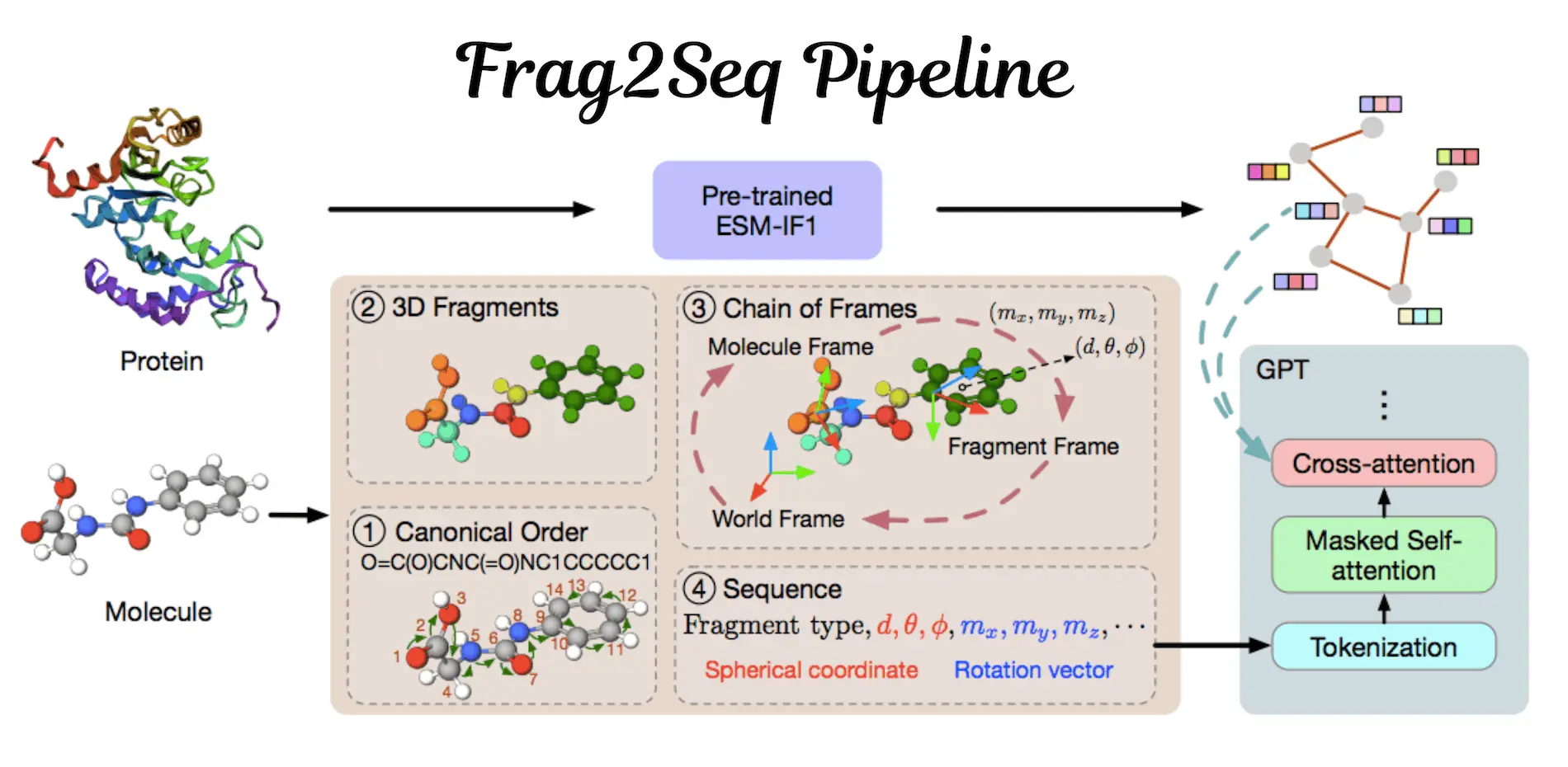
Understanding Frag2Seq
Herein, researchers propose the utilization of LMs to generate molecules for the SBDD problem in a fragment-based manner. This was done through the construction of SE(3)-equivariant molecule and fragment local frames, from which SE(3)-invariant sequences were derived containing the SE(3)-invariant coordinates and orientations of the 3D fragments. This is a new approach for translating the 3D molecules into fragment-informed sequences. The protein pocket embedding is used from an existing inverse folding pre-trained model and cross-attended with LMs to consider protein conditions. Therefore, the approach does not lose geometric information during the sequence conversion and enables using strong LMs to construct target-aware molecules. This is evident from the experimental results, which outperformed strong baselines in efficiency and effectiveness.
Performance of the Model
The relative binding affinity and other pharmacological properties of the produced compounds are better than those obtained by previous baseline methods. That is why an approach helps to achieve the best outcomes on Lipinski and QED, which contributes to enhanced pharmacological properties. Boolean or simple numerical interactions between ligands and protein pockets are represented by achieving the greatest affinity value and the optimal Vina score. The fragment-based method and relatively simple procedure make it easier to sample at a higher efficiency than previous methods. The produced molecules are capable of mimicking the interactions that can exist between proteins and ligands since they give a better binding compared to the reference molecule. The empirical distribution of the created molecules’ and the reference molecules’ carbon-carbon bond lengths are plotted to show how well the technique affords the two modes in the reference distribution. Jensen-Shannon divergence (JSD ) serves as the goal function to make a fair comparison of distribution fitting. The simulation reveals that the proposed method is superior to the existing techniques. In this respect, the produced compounds evidence that the approach can generate new compounds with higher binding affinities while maintaining characteristics comparable to or exceeding a drug.
Conclusion
In this work, researchers introduce a method to convert molecules into sequences invariant to SE(3) movement that the language model can easily comprehend using fragments. To encode protein sequences, researchers use a protein inverse folding model that generates protein embeddings, which they then feed into the decoder via cross-attention.
The application of language models in structure-based drug design is one of the most efficient and effective strategies in enhancing drug discovery, as evidenced by the tests carried out. But of course, there are also some limitations to this study as well. Moreover, real number tokenization requires quantization, which decreases precision to some extent; furthermore, chemical bonds have to be reconstructed by OpenBabel instead of being generated by the language model.
Article Source: Reference Paper
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}