
Deep learning is being utilized in the course of drug development to understand the structure of the protein-ligand complex, utilizing the technique of virtual structure measurement that combines docking and deep learning despite limitations in accuracy and efficiency. Here, researchers demonstrate that a single multi-task geometric deep learning model, LigPose, can accurately handle these two basic tasks. LigPose uses graphs to represent ligand and protein combinations to optimize the three-dimensional structure of complexes. It allows for one-step prediction without the need for docking tools by learning atomic interactions and binding strength. LigPose demonstrates state-of-the-art performance in drug discovery, according to extensive studies, suggesting a viable AI-based drug development pipeline.
Introduction
Small organic molecules (SOM), which make up 72% of FDA-approved medications, are crucial to clinical treatment. Protein-ligand complexes, created when SOM binds to target proteins, show bioactivities and provide direction for the creation of structure-based medicines. Comprehending these complexes at the molecular level facilitates lead optimization and drug screening. Traditional techniques, such as cryo-electron microscopy and X-ray diffraction, need a lot of time and resources.
Molecular docking methods have been widely employed in the past few decades to anticipate nativelike conformations of ligand-binding with protein-binding sites using virtual measurement. These technologies pick a top-scoring posture by ranking binding conformations using a scoring formula. However, the success rate of these technologies is only between 40 and 60 percent. Scholars have endeavored to construct hybrid techniques that leverage deep learning as a more expressive scoring function; nonetheless, these approaches are limited by the sampling time and space of docking tools.
About Deep Learning Models
Deep learning techniques such as AlphaFold, RoseTTAFold, and ESMFold have shown a notable ability to predict protein structures, surpassing the capabilities of earlier techniques. By directly creating protein structures from chemical sequences, these techniques challenge the structure prediction paradigm. They show how deep learning techniques can be used for downstream tasks by accurately predicting the architectures of complexes based on amino acids, such as protein-protein and protein-peptide complexes. Virtual screening and the prediction of protein-ligand complex structures are two applications limited by the fact that these techniques are not specially created for SOM ligands. The need to create a deep learning model that can find protein-ligand pairings and give refined atomic complex structure is, therefore, more suitable.
Understanding LigPose
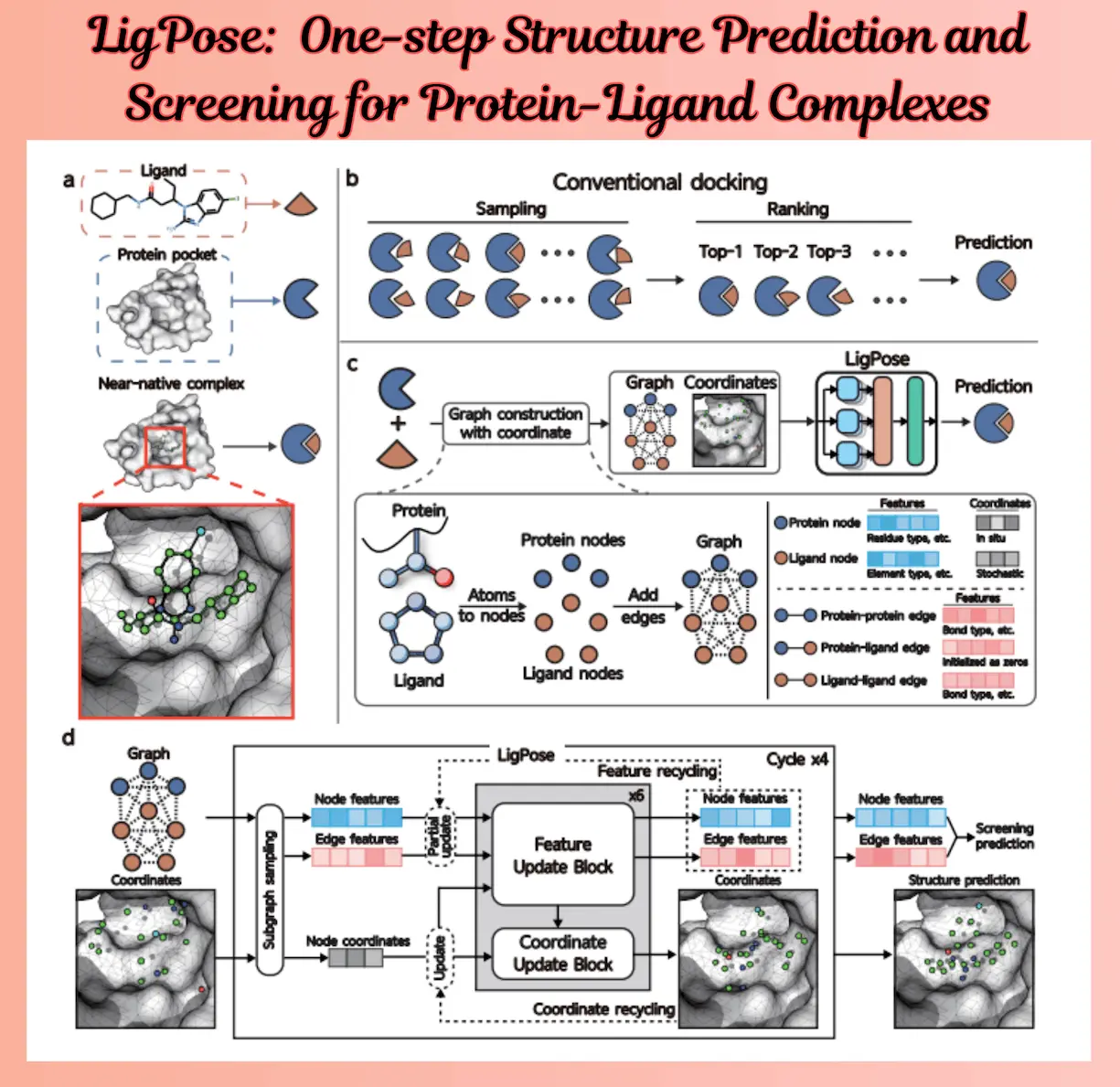
LigPose is a novel geometric deep-learning technique that does not require docking and can effectively predict the native-like shape of ligands with matching protein targets and binding strengths. It entails modeling the ligand and its binding target as an entire undirected graph, where each atom is represented by each node. Atom coordinates in Euclidean space are used to optimize the three-dimensional structures; binding strength and correlation-enhanced graph learning are auxiliary tasks.
It is a deep learning technique that has transformed drug discovery through the use of inter-atom connections for virtual screening, affinity estimate, and structure prediction. It achieved 14% better conformation prediction and 1851x quicker inference speed compared to 12 current docking tools. Additionally, LigPose performed better on virtual screening and cross-docking activities, achieving gains of 19.3% and 20.1%, respectively. The SARS-CoV-2 Mpro complex structure prediction showed its resilience in real-world settings, with improvements of 18.2% above docking tools. LigPose has the potential to be used in structure-based drug design through deep learning because it can learn non-covalent interactions without explicitly adding physical or chemical priors, despite its excellent accuracy and speed.
Applications of LigPose
The high-performance technique LigPose provides a huge number of inferences that are much faster than conventional techniques. Up to 1060 drug-like molecule candidates should be appropriately screened in the real world of drug development. LigPose’s effective end-to-end learning framework presents an alternative to docking-based methods, which suffer from efficiency issues. By merging deep learning with traditional docking tools, LigPose’s technology could speed up drug development.
On the improved set of PDBbind, LigPose performed 14% better than 12 well-known docking tools, correctly predicting 3914 ligand structures with an RMSD < 2Å. On the core set, its performance outperforms hybrid deep learning techniques. Because docking tools have limited sample power, hybrid approaches like DeepDock are unsuitable for use in practical drug screening scenarios. By directly optimizing atom coordinates without the need for energy-based minimizing or sampling, LigPose overcomes these drawbacks and offers a revolutionary computational paradigm for protein-ligand complex structure prediction that is akin to AlphaFold in protein structure prediction. LigPose performs remarkably well at a more difficult job, namely cross-docking, showing a 20.1% increase in success rate when compared to the best-recording deep learning approach, RTMScore. In contrast to earlier approaches (DeepDock, DeepBSP), which primarily exploited the re-docking job, cross-docking is more useful in real-world scenarios.
Conclusion
LigPose is a multi-task geometric deep learning model that simultaneously predicts the affinity, screening probability, and complex structure of proteins and ligands. It directly predicts the complicated structure in three dimensions using a graph transformer network structure. LigPose optimizes learning by managing a high number of protein nodes while minimizing the computing burden through the use of a sampling and recycling technique. Additionally, it creates a symmetric-aware loss function and a particular atom initialization technique for ligands with symmetric structures. The model is trained on all tasks by utilizing large-scale unlabelled protein-ligand pairings for self-supervised learning using a multi-task learning framework. These developments greatly enhance the duties involved in medication development. LigPose may help advance de novo drug design, which may be thought of as future research, by developing innovative end-to-end drug-generation techniques with both precise structural information and reliable bioactivity prediction.
Article Source: Reference Paper | The source code of LigPose is available under an open-source license on GitHub.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}