
The goal of de novo protein design (DNPD) is to build novel protein sequences from scratch without using pre-existing protein templates. Nevertheless, existing deep learning-based DNPD methods are sometimes constrained by their concentration on particular or poorly defined protein designs, which prevents more extensive investigation and the identification of a wide range of useful proteins. In order to solve this problem, scientists from Westlake University present Pinal, a probabilistic sampling technique that produces protein sequences by employing rich natural language as a guide. In order to improve search inside the large sequence space, the work employs a language-based method to construct novel protein structures within a smaller structure space. Tests reveal that Pinal works better than current models and can adapt to new protein configurations, which is beneficial to the biological community.
Introduction
Proteins are essential to life as they are involved in every biological function in living things. Customizing proteins for particular biological or medicinal uses is the goal of protein design. Despite their effectiveness, traditional protein design techniques are frequently constrained by their dependence on pre-existing protein templates and inherent evolutionary restrictions. De novo design, on the other hand, gains from both viewpoints. First, just a small portion of the potential protein landscape has been investigated by nature. Second, the biological traits that evolution has chosen might not match the unique functional needs. By creating completely new proteins with desired shapes and functionalities through de novo design, researchers are able to overcome the limits of conventional approaches.
Key Contributions of the Study
For the first time, scientists argue and show that the large size of the protein sequence space makes it challenging to train end-to-end for text-to-text sequence mapping. Rather, by utilizing the reduced structure space, a two-phase design method places more limitations on the creation of sequences, leading to notably improved outcomes in terms of several protein design metrics.
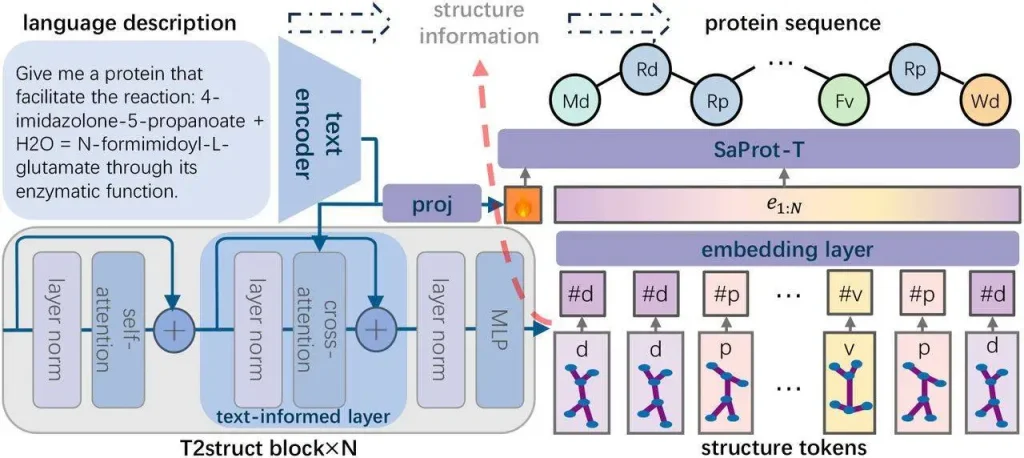
Researchers use deep learning to put the insight into practice and suggest a brand-new DNPD pipeline named Pinal. T2struct and SaProt-T, the two network components that make up Pinal, are responsible for translating natural language into protein structure and carrying out the subsequent sequence design. Furthermore, researchers came up with the best sampling plan to incorporate T2struct and SaProt-T into the Pinal architecture.
Pinal is methodically validated by researchers using a series of studies conducted under both brief and comprehensive educational circumstances. Pinal continuously beats other models, such as earlier techniques and the running ESM3 (Hayes et al., 2024). Additionally, scientists discovered that Pinal exhibits good generalization skills, allowing it to create unique proteins outside the training set.
Understanding Pinal
Pinal is a technique that scientists have created with the goal of designing protein sequences based on the significant correlation between their structure and function. With this approach, sequences are created that are conditioned on the language description and its structure, as well as on the translation of natural language descriptions into structural information. The researchers understand natural language and extract structural information using an encoder-decoder architecture called T2struct. For improved scalability, they additionally employ discrete structure tokens produced by the vector quantization approach. Next, in order to enable sequence creation based on the provided backbone and language instructions, the researchers retrain and adapt SaProt, a structure-aware protein language model, to comprehend natural language inputs. This method guarantees robust foldability and precise formulation of desired functions.
Limitations and Future Research Direction
The functional descriptions of proteins, including amino acids, can be affected by mutation effects. This data is not used for precise design; instead, researchers use it to improve functional descriptions during training. This strategy is difficult since there is a lot of diversity in residue-level data between proteins, and there are no trustworthy techniques to infer the activities of individual amino acids from protein sequences. Consequently, building a robust model requires more than just depending on available data.
The datasets that the researchers used are superior to those in the biological community, but they are still not comparable to datasets in other domains like natural language processing or visual production in terms of quantity or quality. The problem lies in the fact that proteins of interest to biologists frequently have intricate reaction processes yet are not well described, making model building difficult. On the other hand, proteins with a large description are simpler for models to handle, but they have likely previously been thoroughly examined and are not as important. This means that models need to be very good at generalizing, which emphasizes how important it is to have access to better datasets.
Researchers only considered dry experiments for the evaluation in this research. Wet lab trials are how the researchers want to validate the proteins that have been developed in the future.
Conclusion
A unique framework for designing proteins, Pinal is built on natural language. It is converting structural modalities from descriptions of the language used by proteins into sequences that are conditioned on both language and structure. An elegant optimum sampler completes the technique. Pinal proteins beat earlier techniques in a thorough examination of dry experiments, showing great foldability and good alignment with natural language cues. Proteins with previously undiscovered functional combinations can be designed thanks to the method’s ability to generalize beyond training data.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}