

Throughout the years, dissecting gene regulation has been a major challenge for researchers. Using high-throughput sequencing, understanding chromatin landscapes and conformations has set a new era in genomic medicine. Researchers from Shanghai, China, have presented DLoopCaller – a deep learning approach to predict chromatin architecture to identify gene regulation down to transcription factor.
Chromatin is a highly ordered structure containing genetic information (in the form of DNA and RNA) wrapped around histone proteins. These chromatin conformations, or simply the landscape, play a distinctive role in the overall functioning of the cell. Throughout the advancements across new age technologies, understanding these chromatin landscapes has come across as important to dissect 3D structures of the genome and its functionalities. Several technologies, such as Hi-C (High-throughput Chromosome Conformation Capture) and HiCCUPS, have surfaced to capture high-resolution interactions to draw inferences.
Wang et al. and his collaborative team have presented a deep learning approach to predict these chromatin landscapes and loops with improved accuracy and efficacy- DLoopCaller. The research team has evidently shown better result prediction and overall performance in reference to other state of art technologies at the moment, such as Peakachu. Several limitations are associated with earlier technology, such as the inability to predict the impact of structural changes within the genome for disease or gene mutation.
Researchers have defined a working model of DLoopCaller, it is based on a deep learning framework to predict the chromatin loops within genome-wide landscapes using NGS technologies such as Hi-C matrix and available chromatin 3D landscapes. Posing similarities to Peakachu, the deep learning approach uses experimental data from ChIA-PET/HiChIp and Capture Hi-C (positive interactions) to build a binary classification. The classification considers the interactions as positive while non-interactions as negative.
The research team has established its functioning and validated the approach by experimenting on four cell lines chronic myelogenous leukemia, lymphoblastoid cell, hematopoietic stem cell, and mouse embryonic stem cells. The data inputs, including available chromatin landscape data and Hi-C data, were converted to 10kb resolution. The positive interaction verified from Peakachu GitHub was used to produce training samples for model training.
The framework was formed using a three-layer convolutional neural network (CNN) to extract features from the complex database of biological data. Following the route of image processing research, DLoopCaller uses two-channel input into the model. The framework layer is comprised of three convolutional blocks consisting of each layer a dropout layer, a global average pooling layer, and a ReLU layer.
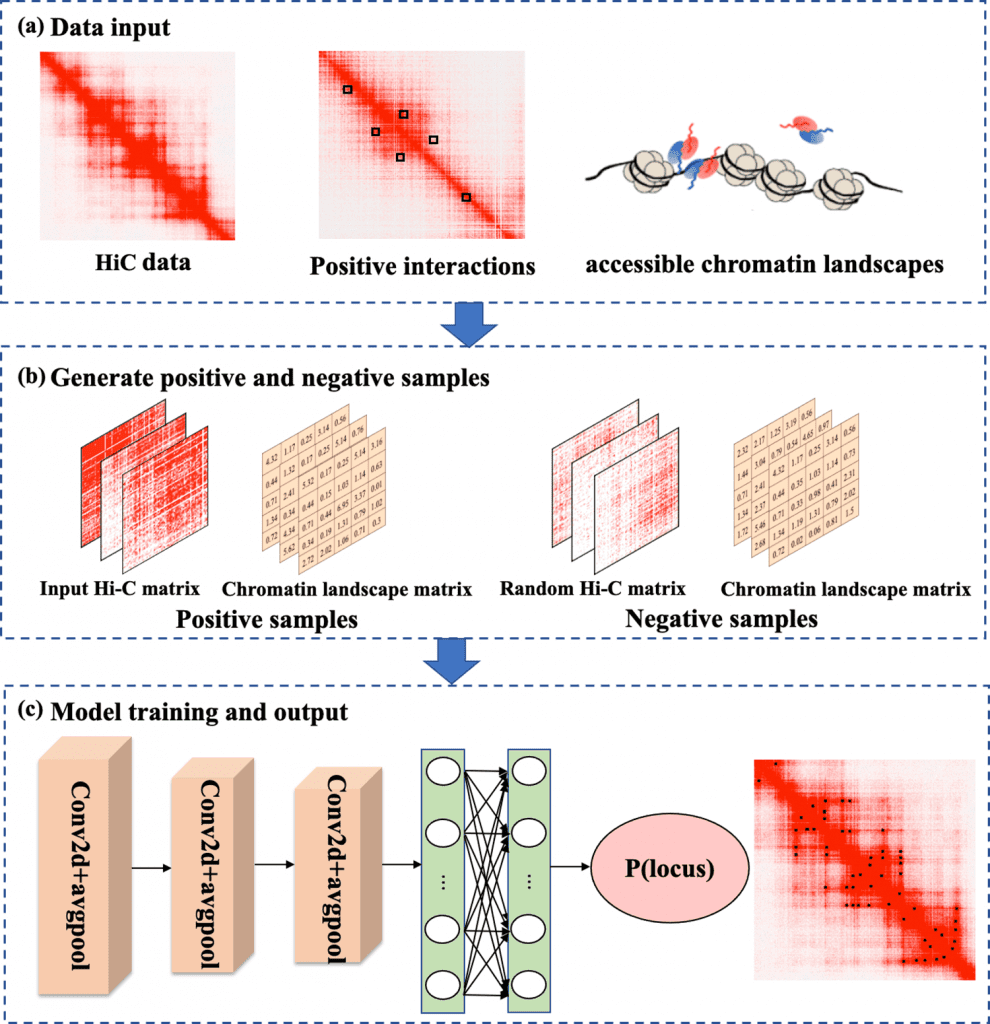
Image Source: https://doi.org/10.1371/journal.pcbi.1010572
Upon model training and identifying genome-wide chromatin loops, by training each chromosome with the model, the prediction process is used for potential loops in identical chromosomes. The overall result for each cell type showed the model’s ability to learn and identify features automatically. As compared to Peakachu, DLoopCaller proved to be outperforming it with the limitation of low resolution. Applying experimental data from each cell type for the predicted loop in the trained model, aggregated peak analysis (APA) was used to quantify the captured loops against Hi-C signals. The resulting peak for each cell line depicted the ability of the deep learning approach to predict transcription-enhancer-regulated chromatin loops. This effectively determined the sensitivity of DLoopCaller to identify genetic aberrations associated with disease genetics.
In order to identify cell type specificity based on chromatin loops, the research team used DLoopCaller to overlap chromatin loops in the three cell lines. Two chromatin loops were considered a match if around 10kb region was found to overlap around the center. The overlapping loops were seen with increased tolerance; hence Wang et al. drew inferences for identified chromatin loops that are highly specific to each cell type, providing distinguished differences.
Earlier results demonstrated the specificity of the model to identify enhancer-promoter interactions. The team attempted to understand the particular transcription factor and their cooperation to determine the cell-specific genomic interactions. Using Spatzie, an R package that identifies significant transcription factors. The correlation was able to show strong interdependence between KLF5 and ZN700, KLF3, KLF6, SP2, and SP1 motifs in H1-ESC, identifying significant transcription factors across different cell lines.
Considering the functionality and specificity of DLoopCaller on the trained dataset, researchers specified major contributions made by deep learning approach such as:
1) Using Chromatin architecture to generate a matrix that matches high throughput Chromosome Conformation Capture (Hi-C), devoid of manual labor of extracting data.
2) Apply massive experimental data from ChIA-PET/HiChIP and Capture Hi-C to identify positive and negative interactions.
3) Constructing a deep learning framework with absolute specificity and extracting features from the Hi-C matrix and available chromatin landscape to identify chromatin loops.
Final Thoughts
DLoopCaller was able to perform with absolute accuracy and help in drawing new inferences from data in comparison to its predecessors, such as Peakachu. But scientists did address the limitations associated with the model, such as the dependence on training data based on experimental enrichment data. Since deep learning poses a black box model, the extracted information does seem difficult to interpret. Hence, the approach can overcome limitations and pursue regulatory identification within genomic data.
Article Source: Reference Paper | Source Codes: DLoopCaller
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.





{kind=link}