
Understanding the complex mechanisms behind many essential biological activities is essential for developing new drugs and has broad ramifications in the disciplines of biotechnology, medicine, and drug development. Still, about 200 million proteins are uncharacterized, and computational attempts foresee annotations of different quality, mostly depending on protein structural information. Here, scientists describe a method for predicting protein activities just based on the sequence by using graph networks informed by statistics. PhiGnet is a quantitative evaluation technique that allows for the quantitative evaluation of particular functions by characterizing evolutionary signatures. Even in the absence of structural information, it reduces the sequence-function gap. In research and biology, this approach identifies functional sites at the residue level, offering significant assistance in the interpretation of the characteristics and novel functions of proteins.
Introduction
The prediction of protein structure has benefited greatly from the evolutionary knowledge found in protein sequences that have been obtained through genome sequencing. Protein functional sites have been characterized by the couplings between paired residues, which capture interactions between residues that support particular functionalities. The identification of disease variations, allosteric mechanisms in proteins, and metamorphism in proteins—reversible transitions between distinct folds, frequently accompanied by diverse functions—have all been made possible by this data. Comprehending the molecular functioning of living creatures, health, sickness, and evolution requires an understanding of protein function.
Approximately 356 million proteins are found in the UniProt database; of these, 80% have no known functional annotations. Due to the length of sequences, traditional techniques of annotating protein functions are constrained, and function annotations are frequently assigned at the protein level. Protein 3D structure prediction has become remarkably accurate thanks to deep learning techniques, which involve millions of parameters and don’t make assumptions about input or output data samples. Still, it is not easy to correctly annotate proteins with functions, particularly when contrasted with experimental findings. In spite of the abundance of available data, recent gains in a variety of domains have been fuelled by physics-based deep learning techniques, which have improved machine learning’s ability to develop interpretable solutions for scientific issues. It is more difficult to assign a protein’s function than to predict its three-dimensional structure, even after decades of devoted research. Because there are few experimentally confirmed protein structures and confidence scores of computationally predicted structures vary, state-of-the-art methods that use structural information have had less success in reliably assigning protein functions. The absence of a quantitative description of residue roles makes it difficult to use a scoring function to evaluate the importance of residues.
Understanding PhiGnet
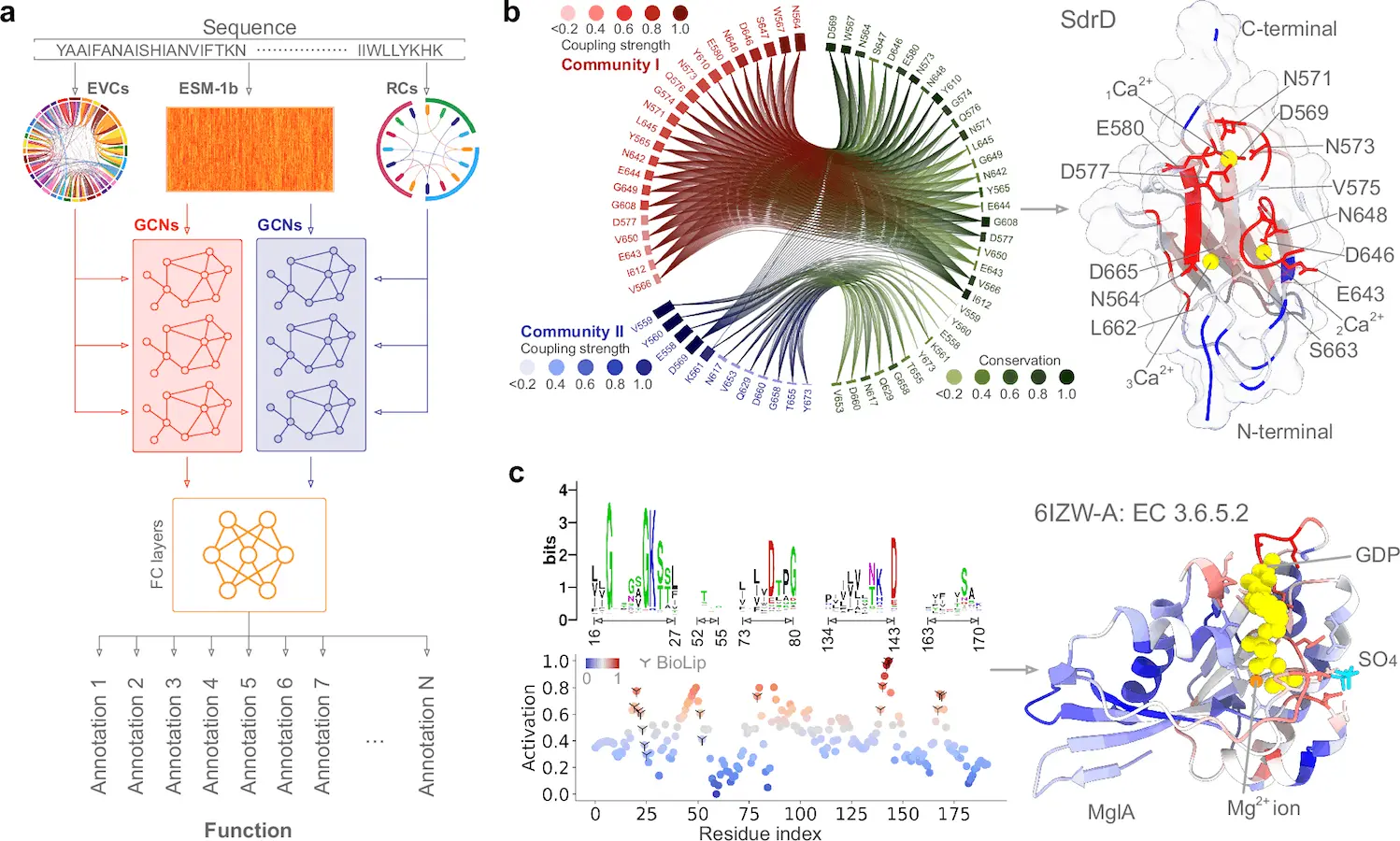
Researchers theorized that they could annotate functions at the residue level by using the information contained in coevolving residues as a means of overcoming these difficulties. Scientists developed a statistics-based learning method called PhiGnet to make protein functional annotation and site identification easier. The technique uses two stacked graph convolutional networks and makes use of the information obtained from evolutionary data. With the help of the developed network architecture and the learned information, the current approach is able to precisely annotate proteins with functions and—more importantly—quantify the relative importance of individual residues to particular functions.
Applications of PhiGnet
In protein function annotations – The PhiGnet technique annotates protein functions and identifies functional locations across species using graph networks influenced by statistics. Utilizing stacked graph convolutional networks (GCNs) in a dual-channel architecture incorporates information from residue communities and evolutionary couplings. Using this strategy, proteins are given functional annotations, such as Gene Ontology (GO) words and Enzyme Commission (EC) numbers. The pre-trained ESM-1b model is used to derive the protein sequence, which is then fed into six graph convolutional layers of dual-stacked GCNs. After analyzing data from the two GCNs, these layers produce a tensor of probabilities that may be used to give the protein functional annotations. The importance of each residue in a given function is evaluated using an activation score.
In protein functional sites – Essential amino acids, which are connected to functional regions such as ligand-binding sites and scattered throughout several structural levels, impact the activity of proteins. A computational technique called PhiGnet can quantify how important each amino acid is for a given function. Nine proteins, including cPLA2α, TpK-BTK, Ribokinase, and HPUK, were used to test the approach. With an average accuracy of 75%, the results demonstrated promising accuracy in predicting important sites at the residue level. PhiGnet reliably finds highly activated residues in proteins that are functionally important.
Conclusion
Using high-order patterns from protein sequences and evolutionary data, the machine learning model PhiGnet has demonstrated better performance in understanding protein function. As a result, the functions of proteins can be understood more fully and precisely. The complex link between protein sequences and their functions is made clear by PhiGnet’s efficient assimilation of increased evolutionary information. In comparison to other methods, it also attains greater accuracy in Fmax, especially when handling proteins with low sequence identity. This indicates how generalizable PhiGnet is in predicting protein function annotation across GO keywords and EC numbers.
Article Source: Reference Paper | The PhiGnet Python code and pre-trained model are available on GitHub
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}