
Scientists at the Massachusetts Institute of Technology and Tufts University introduced a deep-learning model named ConPLex that executes sequence-based predictions of Drug Target Interaction (DTI) with high accuracy, specificity, adaptivity, and alacrity, outperforming other state-of-the-art approaches and unlocks prospects in accelerating drug discovery process by making in-silico screening strategies more viable at massive scale.
Therapeutic Development Demands Advancement In Existing Computational Tools
Multiple Large-scale Genome projects will not succeed in achieving the objective of revolutionizing the healthcare sector with extensive Precision and personalized medicine unless the period and expense of the therapeutic discovery pathway are compressed. Although many facets are yet to decode, multi-omics presented substantial information regarding the progression of lethal disorders like cancer, metabolic diseases, cardiovascular disease, etc. However, the productive drug design phase to remediation is rate-limiting and challenging.
Prescribing medication to patients comes with a tremendously long journey. The drug discovery stage takes decades of research and trial. Screening the interaction of the drug and the target alone is an exhaustive and expensive task. Computational techniques such as Molecular docking, Active site modeling, Rational Design, etc., now routinely assist researchers worldwide in lead generation and comprehending possible Drug Target Interactions (DTI). However, these tools are computationally intensive and don’t fulfill the demand of large-scale screening of every probable target and drug at once directly. Researchers of MIT and Tufts University approached to counteract these significant constraints with anticipation of expediting therapeutic development strategies.
ConPLex: Integrating Contrastive Learning (Con) and Pretrained Lexicographic (PLex)
ConPlex offers sequence-based DTI (Drug Target Interaction) prediction harnessing pretrained Protein Language Models (PLMs) and contrastive learnings. Other methods for sequence-based DTI prediction utilizes Convolutional Neural Network and Transformer, often lack accuracy due to limited DTI training data, and cannot differentiate between decoy compounds or false positives that show the similar physiochemical property but do not bind with the target and true positive.
PLMs can learn the distributional attributes of amino acid sequences over millions of proteins in an unsupervised manner, generating sequence-based representations that encode deep structural insights and thus can overcome the shortcomings of limited DTI data availability. Contrastive learning is a protein-anchored contrastive coembedding that colocates the proteins and the drugs into a shared space. The coembedding facilitates separation between true interacting partners and decoys and thus achieving high specificity. ConPLex leverages advances in PLM to achieve both generalizability and specificity.
The model guided learning by alternating between two objectives over multiple iterations: a coarse-grained objective and a fine-grained objective. The coarse-grained objective trains the model to predict the drug target cognate in the DTI space, and the fine-grained objective adapts the model to distinguish between true and false positive interactions in the DTI space. Makers used Morgan fingerprint and embeddings from a pretrained ProtBert model to featurize the inputs.
Advantageous Elements of ConPLex
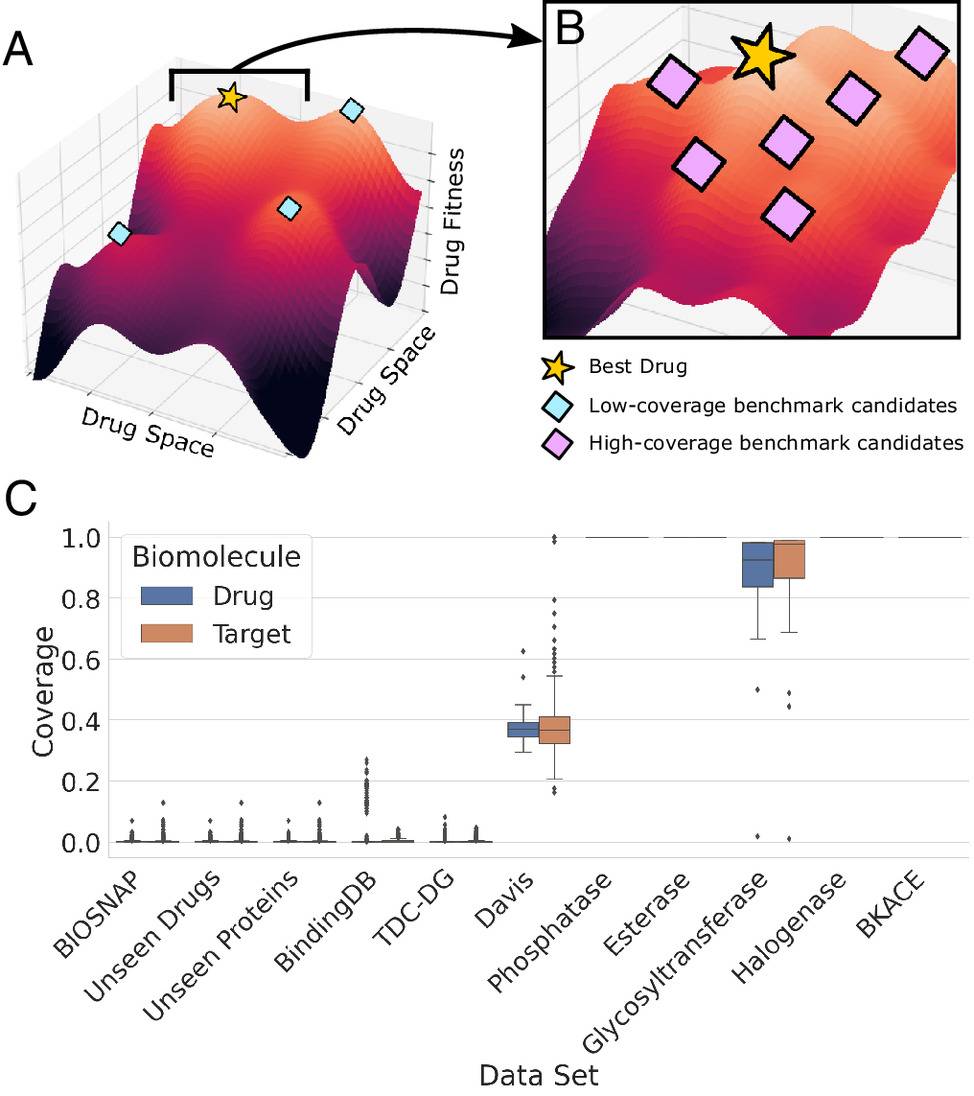
ConPLex performs especially well compared to current methods in the zero-shot predictions where no information is available about the protein or drug during training. ConPLex can also be adapted to predict binding affinity at the subnanomolar level. This was validated by assaying KD values of 19 interactions between 14 compounds and five kinases, and the results correspond with the known findings in the literature. The coembedding of proteins and drugs in the same space delivers better accuracy and interpretability.
Furthermore, the developers made predictions for the human proteome against all drugs in the ChEMBL dataset (2 ×1010 pairs) within 24 hours using an NVIDIA A100 GPU. Such rapid computing speed is an extremely favorable feature for genome-wide screening, massive compound-library screening for drug repurposing, etc. The contemporary approaches also fail to meet this aspect. ConPLex executes state-of-the-art performance on BIOSNAP, BindingDB, and DAVIS datasets and outperforms other state-of-the-art methods- EnzPred-CPI, MolTrans, GNN-CPI, and DeepConv-DTI and single-target Ridge regression model.
Conclusion
Due to its advantageous features of distinguishing decoy compounds, high speed, and generalization of unknown drugs and targets, ConPlex will help researchers speed up screening possible therapeutic candidates. Its attribute of computing large-scale data at genome, proteome, and compound library has the utmost possibilities in improving personalized medicine, drug repurposing, and predicting drug effects against rare variants from underrepresented populations and thus can effectively complement and supplement experimental screening procedures. As a result, researchers are now equipped with an additional tool that holds the potential for saving lives, enhancing their capabilities in this crucial endeavor.
Article Source: Reference Paper | Reference Article
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.





{kind=link}