
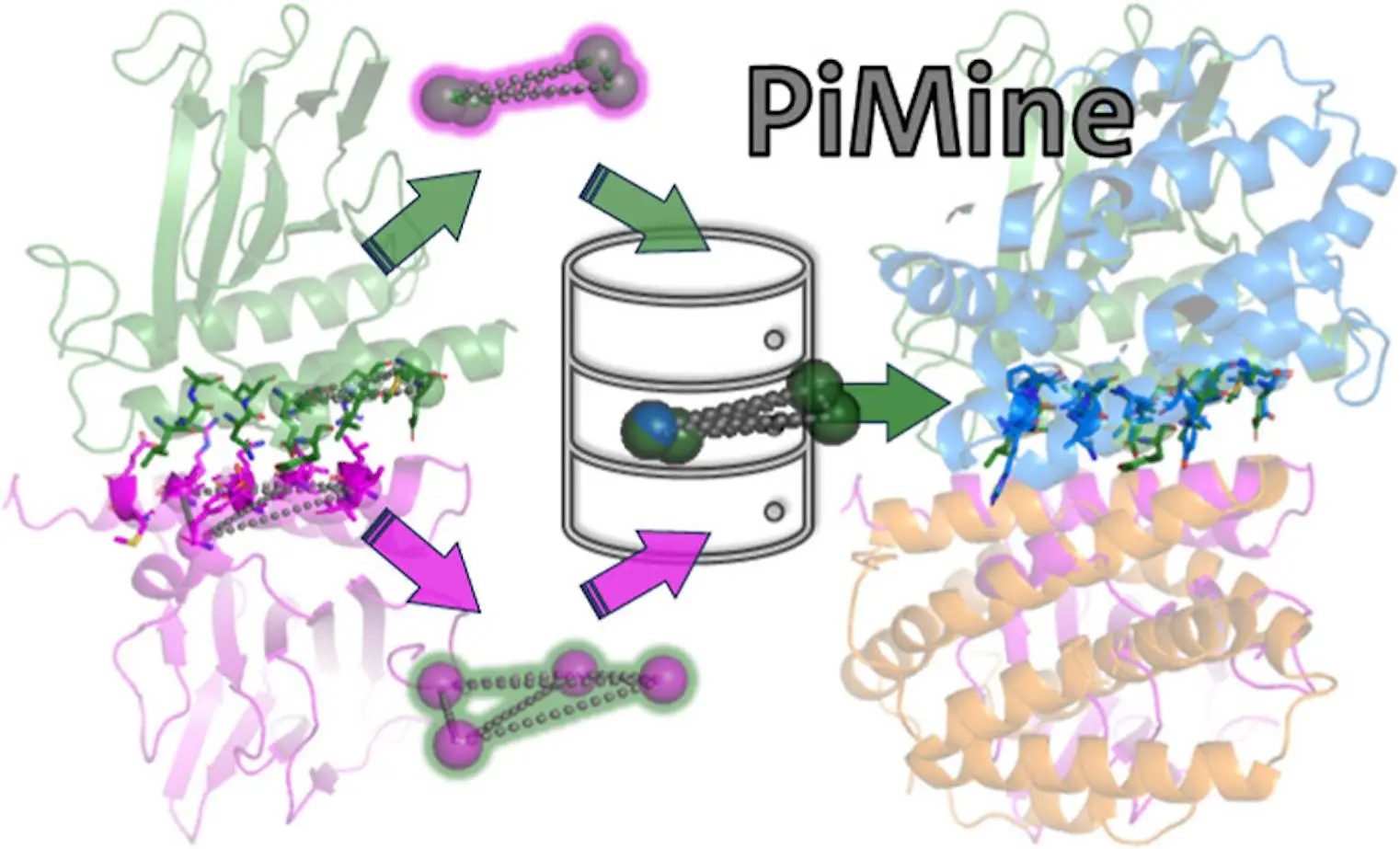
Researchers at Universität Hamburg introduced PiMine, an application that senses structurally related protein-protein interfaces while being driven by a database. PiMine analyzes these interfaces, which are sources of information about protein function and are also important in drug discovery. It distinguishes itself from other programs by the ability to scrutinize interfaces in terms of structural similarity, making it reliable when evaluating interactions between proteins. By exploring known protein-protein interfacial datasets, PiMine can recognize interacting partners as well as possible drug targets.
Unlocking Protein Interaction Potential
Protein-protein interactions are important to unravel the mysteries of many biological processes and key targets for drug discovery. Revolutionary research has recently introduced PiMine, a state-of-the-art approach that uses databases to find similarities between protein-protein structures. The arena of protein interaction studies becomes different with this innovative method and advanced features associated with PiMine. One way to do this is by probing the structural attributes that define interacting proteins within the framework of a given set of protein interfaces. This new development helps us understand more about how proteins interact and also opens up new paths for drug development.
Key components of Database-driven discovery
The structurally comparable protein-protein interactions usually comprise many critical components, such as:
Interface Modeling
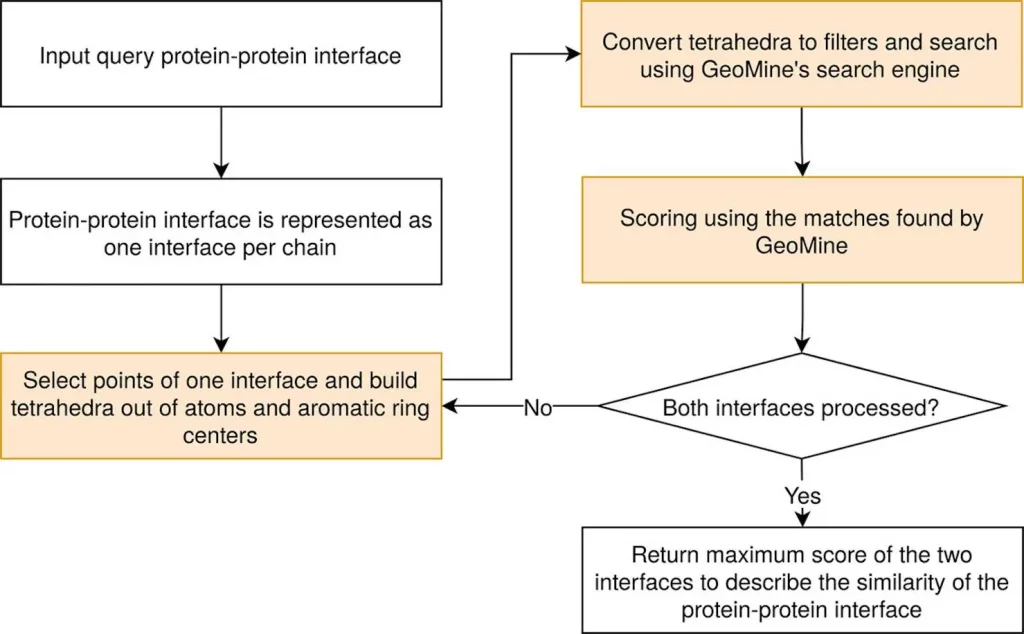
PiMine and other tools of its kind determine protein-protein interface regions by studying the closeness of protein chains. Any heavy atom from one chain must be within 4.5 Å of any heavy atom from another chain to be part of the interface.
Alignment Heuristic
TetraScan is a superior approach used to compare binding sites and protein interfaces. It utilizes tetrahedron-based searches derived from protein site atoms and stored in GeoMine database technology. These searches are compared against PiMine databases that contain interface atoms derived from protein-ligand or protein-protein interactions. This method supports configurable interface requirements, distance range-based atom reduction, distance range-based tetrahedron formation, etc. Filters are thus created using these tetrahedrons to query the PiMine database. The resulting search outcomes exhibit similar patterns to the target interfaces with superimpositions carried out using the Kabsch-Umeyama method. Similarity grooves were obtained by utilizing shape score prefiltering as well as atom-wise scoring methods. For further investigation, this technique calculates all possible chain pair alignments for each similarity in interaction, listing the matches with associated scores and modified target interfaces in pdb format.
Similarity Measure
In PiMine, researchers use three similarity scores: shape score, pharma score, and combined SP score. All the scores are computed for each solvent-exposed site found within the interface using a linear search or k-dimensional tree implementation reliant on the size of the interface. Then, find the adjacent point in the target interface to any query point within a given radius. If the matched point is found at this step, then the shape score rises while the pharma score changes according to pharmacophore attributes with the pre-set matrix. These normalized scores are calculated by dividing them by numerous points that define the surface in the larger two interfaces, giving us shape and pharmacophore scores for each hit. The optimal alignments are those with the highest SP-score against a target interface.
Dataset
Five datasets were used in the studies undertaken. ParamOptSet compared predicted complex shapes to real complexes. It checked how well protein-protein docking scored complex predictions. Dimer597 and Keskin sets used TM-align for interface chain comparisons. PiMineSet evaluated PiMine’s ability to find similarities across single-chain interfaces from different chains. RunTimeSet looked at runtime performance to predict biological protein-protein interactions across diverse protein structures.
External Tools Used
Other tools, such as iAlign and I2I-SiteEngine were used in the study. I2I-SiteEngine was installed and run under default parameters, and the best score ever recorded was used to evaluate its performance. Transformation matrices have been applied using a pdb set from CCP4 Software Suite to target PDB structures as already described above. Version 1.1 of iAlign was executed with settings modified to facilitate protein-peptide interactions properly. The iAlign-IS and iAlign-TM scoring functions were assessed accordingly by taking into account different factors affecting the entire system. However, some problems did occur, like when lowercase chain IDs involved an incorrect identification of some interfaces in RunTimeSet, which otherwise could have been correctly recognized.
Application and Impact
Database-driven algorithms used to identify protein interactions based on structural similarities have significant implications:
- Evolutionary Insights: PiMine’s ability to detect similarities in protein structures across different species offers unique insights into protein function and evolution
- Drug Discovery: PiMine’s impact on drug discovery spans target identification, drug design, and optimization.
- Protein Function Annotation: PiMine’s structural similarity calculations and database-driven methods provide valuable information about the involvement of proteins in cellular processes.
- Protein Engineering: By leveraging structural similarities, PiMine aids in the design and engineering of proteins for desired functions.
Conclusion
PiMine is considered a novel, sequence-independent method for interface matching and alignment of protein-protein interfaces. It competently detects the similarities existing in the interfaces of protein chains not evolving from a mutual ancestor in the PDB database and performs substantially better than I2I-SiteEngine in all respects. Although iAlign is faster, PiMine is more efficient in discovering similarities between sequentially distant yet structurally analogous interface pairs.
Finally, PiMine’s ability to assess individual scores for single interface pairings without the conformation of two chains can help in modeling protein-protein complex architectures, identifying yet-to-be-discovered binding partners, and predicting potential small molecule ligands. This is particularly useful when you only have one interface partner available to search. The tool’s usability has been validated by retrospective and prospective experiences. Screening is recommended using the runtime-optimized options, while the accuracy mode is best reserved for deep similarity analysis. PiMine is distinct in that it lets you utilize single-chain interfaces that are prepared by external programs for database scanning. Predicted improvements could include additional statistics on match significance and searching on protein surfaces ranging from local to global. Overall, PiMine is a useful technique for better-interpreting protein-protein interfaces at the molecular level.
Article Source: Reference Paper | PiMine can be accessed at the Website
Follow Us!
Learn More:




Anshika is a consulting scientific writing intern at CBIRT with a strong passion for drug discovery and design. Currently pursuing a BTech in Biotechnology, she endeavors to unite her proficiency in technology with her biological aspirations. Anshika is deeply interested in structural bioinformatics and computational biology. She is committed to simplifying complex scientific concepts, ensuring they are understandable to a wide range of audiences through her writing.











{kind=link}