
An increasing number of studies employ metagenomic analyses to investigate the taxonomy and function of microorganism communities. In many microbial research, one typical job is to retrieve metagenome-assembled genomes (MAGs) by binning contigs from metagenomic data. Yet, during this procedure, a substantial amount of information is lost. It is, therefore, essential to have functional and taxonomic matrices for each contig. Now available for additional research are Pacbio HiFi reads, a lengthy and superior substitute for short Illumina reads. To analyze both short and long reads, researchers from Université de Toulouse created a workflow that can be easily installed and used on a computing cluster. The workflow can be configured to analyze at the contig or bin level, depending on the user’s preference, and depends on singularity images to remedy dependencies. MetagWGS is a Nextflow workflow focusing on binning (long and short reads) and HiFi read analysis. It has two singularity pictures. Its capabilities and ease of installation and use are demonstrated by comparison with PacBio’s MAG building methodology and a publicly available dataset.
Introduction
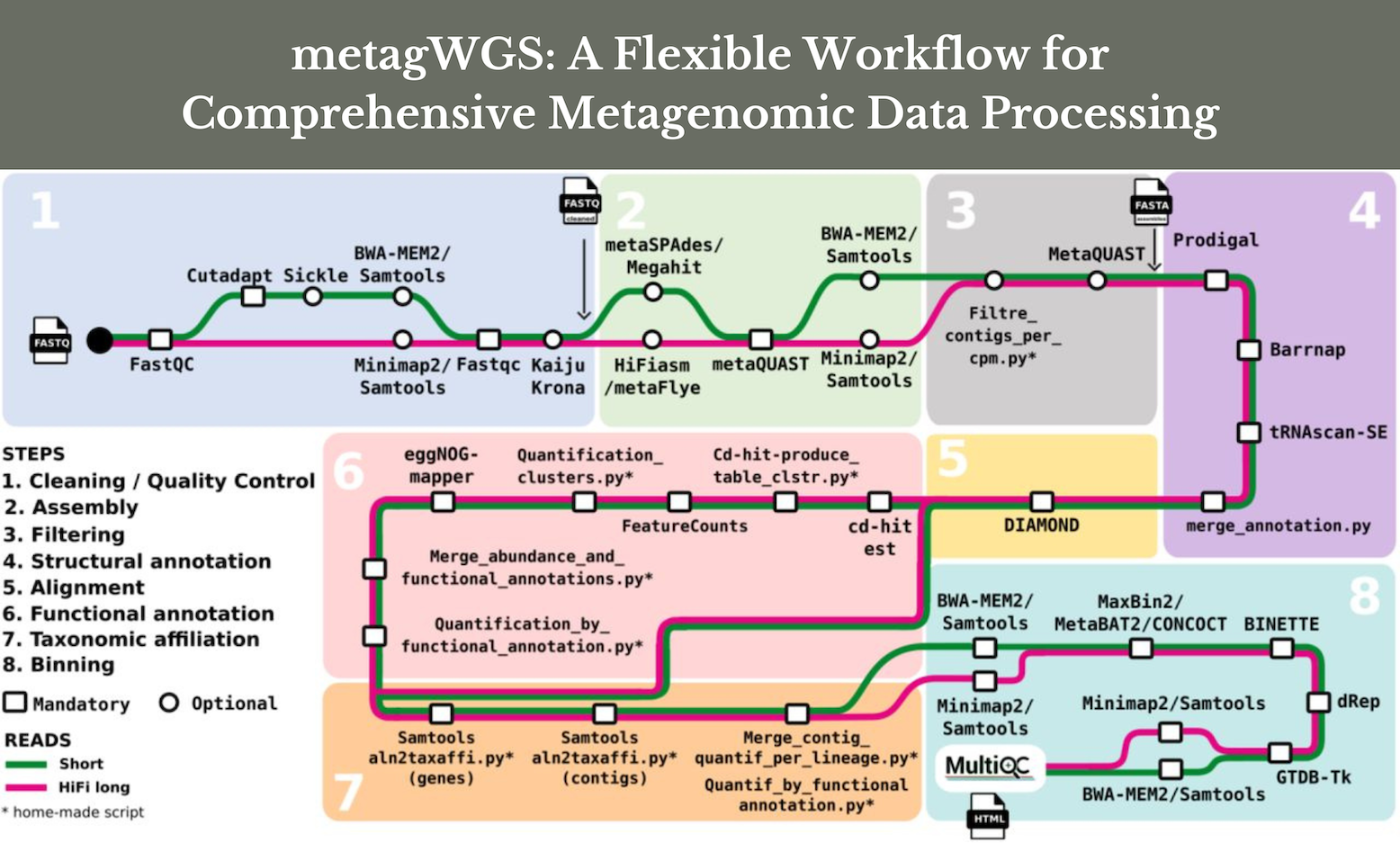
The taxonomic and functional diversity of the communities under study can be accessed by metagenomic analysis, allowing for the reconstitution of the genomes of the most easily assembleable species. Some strategies concentrate only on reads, contigs, and MAGs. A reference catalog’s homology searches work well for situations that have been thoroughly explored. A de novo strategy is advised for communities that have received little research. Taxonomic and functional abundance matrices from as many contigs as possible are created and then sorted into bins in order to gather as much information as possible. The abundance of MAGs (metagenome-assembled genomes) in every sample is determined. All necessary steps are performed by installing and running a single tool, which also conducts binning, dereplication of bins, cleaning, quality checking of the data, assembly, mapping, quantification of contig abundance, taxonomic and functional annotation, functional abundance matrix construction, and building of MAG abundance matrices.
Though they are not intended to produce taxonomic and functional profiles for every contig, existing tools like MAG, MetaWRAP, Veba, and Atlas are meant to be used for reconstructing and analyzing MAGs. Tools such as Veba and MetaWRAP from Uritskiy can provide these profiles, but they are not part of a workflow that is ready to do the whole study on computing clusters.
What is MetagWGS?
MetagWGS is an adaptable assembly tool that combines different binning algorithms with a novel bin refining tool called “Binette” to handle short reads (Illumina) or PacBio HiFi reads. It provides taxonomic and functional annotation for all genes, generates contigs and bins, and delivers high-quality genome bins. Compared to the Pacbio HiFi specialized approach, MetagWGS generates more medium and high-quality bins on 11 public metagenomic samples based on human gut data.
Complete shotgun sequence metagenomic data, such as PacBio HiFi reads and Illumina short reads, can be analyzed using the Nextflow DSL2 workflow. Contigs, genes, and metagenome-assembled genomes (MAGs) are all analyzed by the thorough MetagWGS system. With contigs and MAGs, it generates a taxonomic abundance table; with the catalog of genes present in contigs, it generates a functional abundance table; and Binette, an enhanced algorithm for automatic bin refinement, is the result.
A wide range of biological issues in shotgun metagenomic research are addressed by the pipeline, which finds a balance between accessibility and versatility, enabling users to customize their analyses. Researchers used a public dataset of 11 human gut metagenomic samples to compare metagWGS with the HiFi-MAGS-pipeline, a MAG-building technique proposed by PacBio.
Future Direction
Researchers plan to investigate the use of co-binning as an alternative to cross-alignment, where reads are aligned on the concatenation of all assemblies. This method offers a consistent alignment and less parallelization, but it may not be optimal for cluster execution. However, the larger genome size makes aligners perform better in cobinning. Co-binning also presents challenges with redundancy in concatenated assemblies, but it remains an interesting approach to test and compare. Co-assembly binning is an excellent alternative to reduce redundancy, but it may not always be feasible for large, diverse samples. To better consider viruses and small eukaryotes, researchers plan to classify contigs into eukaryotic, prokaryotic, and viral contigs, adapt tools for structural and functional annotation, select adapted binners, and assess bins/MAGs quality. The goal is to improve workflow efficiency and reduce resource usage without compromising results quality.
Conclusion
At present, metagWGS is the only workflow that offers taxonomic affiliation of MAGs, taxonomic affiliation of all contigs (including unbinned ones), and taxonomic affiliation and functional annotation of genes. Moreover, it is the only comprehensive procedure available in PacBio HiFi for evaluating metagenomic sequencing data. More medium- and high-quality bins are produced by metagWGS than by the Pacbio HiFi-specific binning method. It does, in fact, use Binette, which outperforms both the DAS Tool and the metaWRAP bin_refinement step in terms of performance (producing more high-quality bins).
Article Source: Reference Paper | metagWGS is publicly available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}