

The Children’s Hospital of Philadelphia (CHOP) scientists have created ESPRESSO, a new computational tool for analyzing RNA-seq data. ESPRESSO aims to improve the detection and quantification of transcript isoforms, which are variations of a gene that can serve different functions. ESPRESSO is particularly useful when dealing with long-read RNA-seq data, which can be prone to errors due to the sequencing technology used. Researchers interested in studying the RNA repertoire of eukaryotic transcriptomes will find the tool useful.
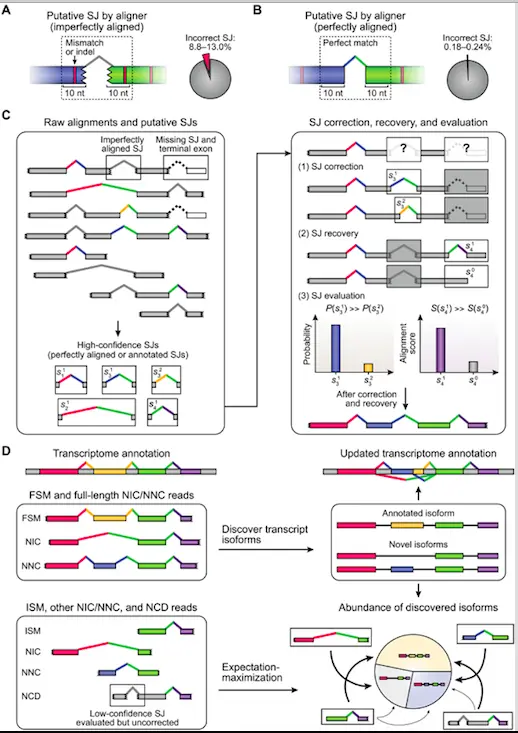
Image Source: DOI: 10.1126/sciadv.abq507
When our cells transform genes into proteins, they frequently pause for a process known as alternative splicing. This is where they take a newly formed RNA molecule and snip and stitch it together in various ways. This can result in one gene producing multiple proteins. This is a normal process that occurs in many biological events, such as when stem cells differentiate into specific tissue cells. However, when things go wrong, and alternative splicing becomes dysregulated, it can contribute to diseases. To get to the bottom of these conditions, it’s critical to examine the entire set of RNA molecules that come from our genes, known as the transcriptome.
Scientists have long struggled to fully “read” RNA molecules because they are typically thousands of bases long. To address this, researchers have traditionally used short-read RNA sequencing, which breaks down RNA molecules and sequences them in much smaller chunks – typically 200 to 600 bases.
The full sequences of the RNA molecules are then reconstructed using computer programs. While short-read RNA sequencing is highly accurate, with a low per-base error rate, it does have limitations due to the short length of the sequencing reads. In other words, it’s like attempting to assemble a jigsaw puzzle with identically shaped and sized pieces but with a much larger picture.
ESPRESSO is a new tool designed to address the challenges of error-prone long-read RNA-seq data. ESPRESSO is a robust and efficient algorithm for discovering and quantifying transcript isoforms, allowing for accurate identification and characterization of alternative splicing events.
Scientists can use this method to sequence RNA molecules with more than 10,000 bases without breaking them up. However, this method has a disadvantage in that it produces a much higher per-base error rate, typically between 5% and 20%. This high error rate has hampered the widespread adoption of long-read RNA sequencing. It makes it difficult to confirm the presence of new, unknown RNA molecules found in specific conditions or diseases.
How ESPRESSO Works?
ESPRESSO consists of three major steps:
- Align raw RNA-seq reads to the reference genome and detect putative splice junctions (SJs), defining high-confidence SJs based on annotation and dinucleotide motifs and validating with synthetic transcripts.
- ESPRESSO determines the most optimal set of SJs for each read by realigning to high-confidence SJs and selecting the most likely option using a probability calculated from alignment and error profile, also able to recover missing SJs.
- Classify long reads into categories based on the annotation status of SJs in the existing catalog and discover transcript isoforms by collapsing full-length reads into unique chains of high-confidence SJs, quantifying individual isoform abundances using expectation-maximization (EM) algorithm.
Validation and Comparison to Other Methods
The study found that ESPRESSO is a superior tool for discovering and quantifying RNA isoforms compared to other existing methods. They tested the tool using simulated data and real biological samples and were able to generate and analyze over 1.1 billion long RNA sequencing reads across various human tissue types and cell lines, providing valuable insight into human transcriptome variation at the level of full-length RNA isoforms.
Conclusion
ESPRESSO is a powerful and efficient tool for detecting and quantifying transcript isoforms in long-read RNA-seq data that contains errors. The unique algorithm accurately identifies and characterizes alternative splicing events in complex or poorly annotated genomes. Overall, with its proven performance:
- ESPRESSO is a new computational tool for analyzing RNA-seq data that improves the discovery and quantification of transcript isoforms.
- ESPRESSO is particularly useful for dealing with long-read RNA-seq data, which is often error-prone due to the nature of the sequencing technology.
- ESPRESSO can accurately discover and quantify different RNA molecules from the same gene, known as RNA isoforms, using only error-prone long-read RNA sequencing data.
- Results of comparison with existing tools indicate that ESPRESSO outperforms other methods in terms of both transcript discovery and quantification accuracy.
- ESPRESSO is a valuable tool for researchers to study the RNA repertoire of cells in various biomedical and clinical settings.
Article Source: Reference Paper | Reference Article | ESPRESSO
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.





{kind=link}