
Large Language Models (LLMs) have greatly revolutionized text generation and comprehension in the world of Natural Language Processing. Nowadays, they can be used for gene analysis, too, but their knowledge is limited. Enhancing LLMs with external gene knowledge is challenging due to high costs and computational resources. Retrieval-Augmented Generation allows the dynamic use of external information to improve the accuracy of answers. Researchers from The University of Hong Kong, Nanyang Technological University, and The University of New South Wales have developed GeneRAG, a framework that enhances LLMs’ gene-related capabilities using RAG and the Maximal Marginal Relevance algorithm, which outperformed popular LLMs such as GPT-4.
Why aren’t LLMs Enough?
Samuel panics as he tries to wrap up his genetics project.
“What are the top 4 tissues CFTR is expressed in at the highest level?”
After his mom replies, she smiles. “It’ll be alright. You’ll get an A.”
The next day, he comes home, furious. “I got a B, Mom. Prof said my content was wrong.”
“But I used ChatGPT!” she exclaimed while her son groaned. “It hallucinates!”
LLMs struggle with answering gene-related questions due to the complexity of genetic knowledge, which is challenging to encompass within pre-trained parameters.
Existing approaches grapple with enhancing LLMs with external genetic knowledge due to high computational resources and financial costs, making fine-tuning challenging.
Some studies have tried training new models directly using gene information, which proves to be time-consuming and expensive, while still facing integration challenges with extensive external knowledge. This is where Retrieval-Augmented Generation (RAG) comes in. It combines LLMs with retrieval-based methods to improve accuracy by integrating external knowledge, especially useful for gene-related inquiries.
Introduction to GeneRAG and its Methodology
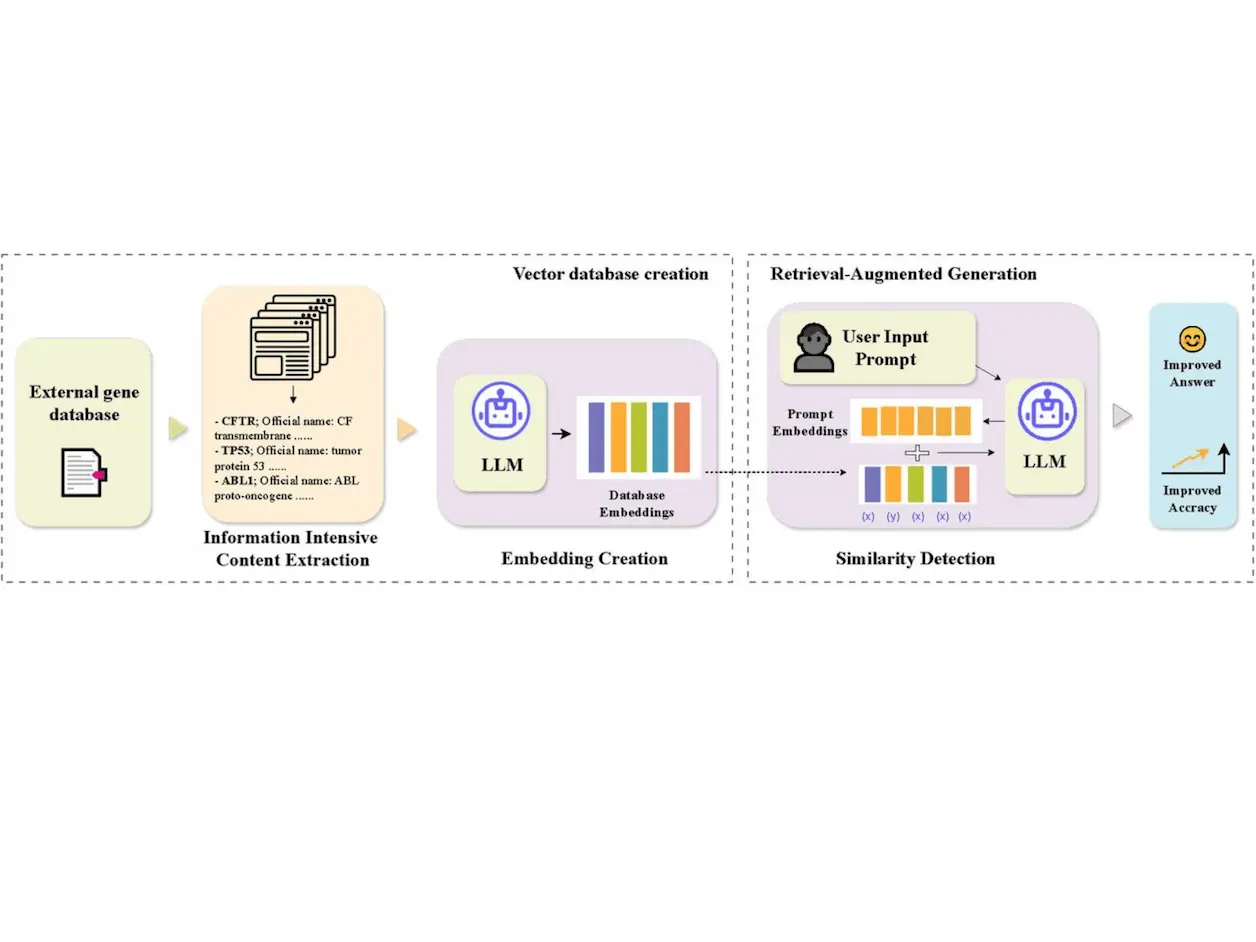
GeneRAG is proposed to be able to integrate external genetic knowledge into LLMs via RAG. First, we discuss the database chosen for this study.
Data Extraction
The NCBI database is chosen due to its credibility and the depth of gene-related data available. In NLP, words are represented in the form of numeric vectors called embeddings. For accurate embeddings, researchers preprocessed the data by normalizing the text, removing duplicates, and ensuring consistency in gene terminology. This preprocessing ensures that the information fed into the LLM is clean and standardized.
They chose an LLM similar to text-embedding-3-large for its ability to understand and generate text that resembles human-like language. This LLM creates meaningful embeddings.
Handling User Prompts
GeneRAG transforms user prompts into embeddings to facilitate similarity detection. Cosine Similarity is used to accurately match user queries with the most relevant gene information in the database. Cosine similarity measures the cosine of the angle between two vectors. Here, they are the embeddings of the prompt and the gene data. The next step is to use RAG.
Retrieval-Augmented Generation
GeneRAG uses the MMR algorithm to balance relevance and diversity in retrieved results for accurate answers. MMR selects documents that are relevant to the query and diverse, ensuring comprehensive and precise information in the final answers. The evaluation metrics are discussed below.
Evaluation and Performance
The researchers answered these questions in the context of GeneRAG’s effectiveness.
RQ1: (Q&A Effectiveness): How effectively does GeneRAG answer gene-related questions?
RQ2: (Downstream Task Effectiveness) How effectively does GeneRAG perform gene-oriented downstream analysis?
RAG Dataset: Contains gene information of 20,350 Homo sapiens genes from NCBI.
Q&A Dataset: Comprises 9,000 questions from the RAG Dataset across three question types.
Downstream Task Dataset: Evaluates cell type annotation and gene interaction prediction using 3,000 cells and genes from two datasets.
Metrics Utilized
Accuracy: Measures the proportion of correct predictions made by GeneRAG.
False Positive Rate (FPR): Indicates the proportion of incorrect positive predictions among all actual negative instances.
False Negative Rate (FNR): Measures the proportion of incorrect negative predictions among all actual positive instances.
Evaluation
For RQ1 tasks, the FPRs and FNRs were lower in GeneRAG than both GPT-3.5 and GPT-4. The overall accuracy of GeneRAG (0.815) surpasses GPT-3.5 (0.594) and GPT-4 (0.722). On average, GeneRAG demonstrates a 39% improvement compared to GPT-4o. Notably, there is a significant decrease in the false negative rate for trap questions, reduced by 65%.
For RQ2 tasks, the Cell Type Annotation Scores of GeneRAG surpass GPT-3.5 and GPT-4 by 66% and 41% respectively.
Conclusion
GeneRAG, leveraging the MMR algorithm and external knowledge bases, outperforms GPT-3.5 and GPT-4 in tasks like answering gene-related questions, cell type annotation, and gene interaction prediction. However, its reliance on external data poses limitations to its scalability and generalizability. Overall, GeneRAG manages to bridge the gap between external information and LLMs’ trained knowledge and provide factual, verified answers on genetics to users.
Article Source: Reference Paper | GeneRAG is available on the Website.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Neermita Bhattacharya is a consulting Scientific Content Writing Intern at CBIRT. She is pursuing B.Tech in computer science from IIT Jodhpur. She has a niche interest in the amalgamation of biological concepts and computer science and wishes to pursue higher studies in related fields. She has quite a bunch of hobbies- swimming, dancing ballet, playing the violin, guitar, ukulele, singing, drawing and painting, reading novels, playing indie videogames and writing short stories. She is excited to delve deeper into the fields of bioinformatics, genetics and computational biology and possibly help the world through research!











{kind=link}