
To deeply understand how tissues work in healthy and diseased conditions, it is imperative to collect vast amounts of data on individual cells. Such data obtained from various studies can then be combined to generate big reference maps of different tissues, helping us to gain clarity on their functions. But, other researchers use different terminologies to describe the same types of cells, and this introduces difficulties in creating a common understanding of cells. To resolve this issue, a group of scientists has devised a novel method called ‘treeArches’, which builds reference maps along with organizing cells into categories that everyone can agree on. It creates a universal language for talking about cells to understand them without conflicts among different research groups.
When the Same Cells Have Different names
Do you know why binomial nomenclature was devised for naming organisms? Because different people call the same animal or plant by different names in different regions, which creates confusion and conflict, especially in biological research. Hence, a universal system of nomenclature was required. Similar is the case of annotations in single-cell sequencing.
Single-cell sequencing has been instrumental in gaining deeper insights into human health. Huge databases called “reference atlases” store single-cell sequencing data for the convenient study of various types of cells in organs which will ultimately help in deciphering the association of particular cells with particular diseases and finding potential treatments. But, creating these atlases is a hassle in itself because the same cell types can have varying names and resolutions across various databases.
That makes it difficult to compare findings from different studies and creates conflicts. To counter this problem, a method called scHPL was developed to build a hierarchy of distinct cell types. Yet, the problem of data privacy still persisted. To put an end to all these persisting challenges, researcher Ahmed Mahfouz and his team have come up with treeArches which builds as well as updates reference atlases and hierarchies of cell types.
treeArches enables scientists to employ existing methods for the creation of atlases, after which scHPL can be used to expand the atlases and highlight the relationships between cell types. Afterward, scientists can analyze new datasets and either derive new insights or utilize the atlases in identifying new cell types in the dataset.
How treeArches Helps Cells Claim Their Identities:
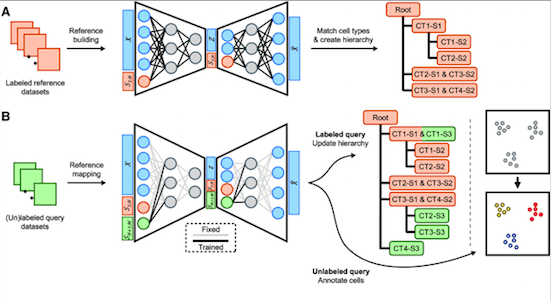
treeArches itself is built around two other tools called scArches, and scHPL, which help it compare various databases of cells and decipher how they form interconnections. The mechanism of treeArches has two main steps:
Step 1: Removing Differences Between Datasets
Scientists take numerous labeled datasets (reference datasets) showcasing different cell types. Then they administer advanced computer models to generate a hidden space representing the similarities between the datasets.
Step 2: Building a Cell-Type Hierarchy
In the next step, scHPL is employed to create a hierarchy of cell types on the basis of the labeled datasets. This hierarchy is similar to a family tree as it demonstrates the interrelationships between multiple cell types.
Now, this hierarchy can be used to examine new datasets, also called query datasets, in two ways:
- The hierarchy can be updated if cell types in the new datasets match those in the reference datasets.
- In case the new dataset is unlabeled, the hierarchy can be utilized to figure out the cell types in the new dataset.
Scientists made improvements in scHPL so that it performs better in integration with treeArches. The scHPL applies a rejection strategy to identify new cells not found in the reference datasets on the basis of specific criteria. There are three methods that aid in the detection of new or diseased cell types, two of which were already present in the earlier version of scHPL:
- Posterior Probability Threshold: The default threshold for this method is 0.5. If a cell shows any lower probability of corresponding to a certain cell type, it may be a new or diseased cell type.
- Reconstruction Error Threshold: This method utilizes the nested cross-validation loop to determine a suitable threshold by considering the median of all the reconstruction errors from the test folds. It helps avoid false negatives (cells wrongly rejected as new cell types).
- Distance Threshold: In this method, first, the average distance between cells and their neighbors in the training set is calculated. The threshold is then set based on the 99th percentile of these distances, and any cell whose distance to its predicted class is way too larger than the threshold is considered to belong to a new or diseased cell type.
For comparing and analyzing the data involved in this process, multiple datasets are used. They are:
- PBMC Datasets:- They contain cells from the bone marrow and peripheral blood mononuclear cells (PBMC).
- Brain Datasets:- They contain genetic information on cells in the primary motor cortex of three species, human, mouse, and marmoset.
- Human Lung Cell Atlas:- It is a reference atlas for lung cells, including the ones involved in the disease idiopathic pulmonary fibrosis (IPF).
Then, methods were used along with treeArches to compare and match cell types between datasets. The methods are listed below:
- FR-Match: It uses marker genes to match cell types in different datasets.
- MetaNeighbor: It compares cell types on the basis of AUROC scores.
- Azimuth: It utilizes the Seurat tool for integrating and mapping different datasets.
Let’s Take a Glance at the Performance of treeArches
At first, treeArches was tested on PMBC datasets to recreate cellular hierarchy matching the manually created one. The results tell that treeArches was indeed successful in matching cell types and recreating the hierarchy. It not only correctly identified cell types shared among datasets but also added new cell types specific to each dataset. treeArches’ performance was compared to FR-Match, MetaNeighbor, and Azimuth and it managed to outperform them in most of the cases except for some with already harmonized dataset labels which fared better with Azimuth.
Then treeArches was used to improve the resolution of HCLA. treeArches updated the cell-type hierarchy using a new dataset called Meyer, and many cell types from the new dataset matched well with the existing ones in the reference atlas. However, there were some mismatches that could be attributed to recent discoveries or differences in annotations. A query dataset called Tata was employed to test the performance of the updated hierarchy, and the results did confirm improved resolution.
In the case of IPF, treeArches was used to detect previously unseen cell types in samples obtained from patients. The original annotations of the IPF cells indicated only broad categorization of the cells. So, scHPL was used to predict more specific cell types for IPF, and the predictions were compared with the original annotations. The comparison revealed that many of the macrophages and epithelial cells in the IPF datasets were rejected, i.e., they were not matched with any cell type on the reference atlas. Upon closer inspection of the rejected cells, it was found that specific subtypes of IPF macrophages were involved in the upregulation of the SPP1 gene, which is associated with IPF. This indicates that the rejected cells could very well belong to the diseased subpopulations.
Apart from the aforementioned cases, treeArches was also implemented in unmasking how different brain cells in humans, mice, and marmosets were related to each other, with an emphasis on GABAergic neurons. At first, data from mice and marmosets were combined to form a hierarchy of brain cell types. Upon comparison, most of the cells between the two species matched, but ‘Meis2’ and ‘Sncg’ showed a mismatch suggesting specificity to species. The hierarchy resulting from combining data on human brain cells was logical and, as expected, according to the names of the cell types.
Conclusion
There are uncountable types of cells in the bodies of living organisms. To study these cells conveniently, it is essential that each cell type has a particular name and information associated with it so that there is no confusion resulting from overlapping data. Since it is possible that different research groups can give the same name to distinct cell types, a method such as treeArches can be beneficial to incorporate to avoid conflicting information. treeArches has shown its efficiency in matching cells to their respective cell types and creating cellular hierarchies that everyone can agree upon. It also demonstrates the ability to detect new cell types that were not observed before. However, a major limitation is the possibility of inaccuracy if the original annotation is erroneous. But, for the most part, treeArches offers a robust analysis of reference datasets, and a few more improvements can overcome its limitations, boosting its efficiency considerably.
Story Source: Reference Paper | Code Availability GitHub
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.





{kind=link}