
The field of protein modeling has witnessed remarkable advances in recent years, particularly with the advent of machine learning techniques. A new review by the Massachusetts Institute of Technology, Cambridge researchers covers the basics of diffusion models, molecular representations, generation capabilities, prevailing heuristics, limitations, and forthcoming refinements in computational structural biology. Among these, diffusion models have emerged as a powerful tool, revolutionizing our approach to understanding protein structures and their interactions.
Diffusion models (DM) are a family of probabilistic generative models that aim at cleaning up tainted samples by making gradual changes. Originally developed under the influence of nonequilibrium statistical mechanics, DMs became rather successful and now are the state-of-the-art methodology in many domains, such as computer vision. These models use stochastic differential equations (SDEs) to model forward and reverse procedures where noisy samples are gradually improved in the direction of the desired data distribution. In the context of protein structure and docking applications, understanding the technical principles and advancements of DMs is crucial for leveraging their potential in computational structural biology.
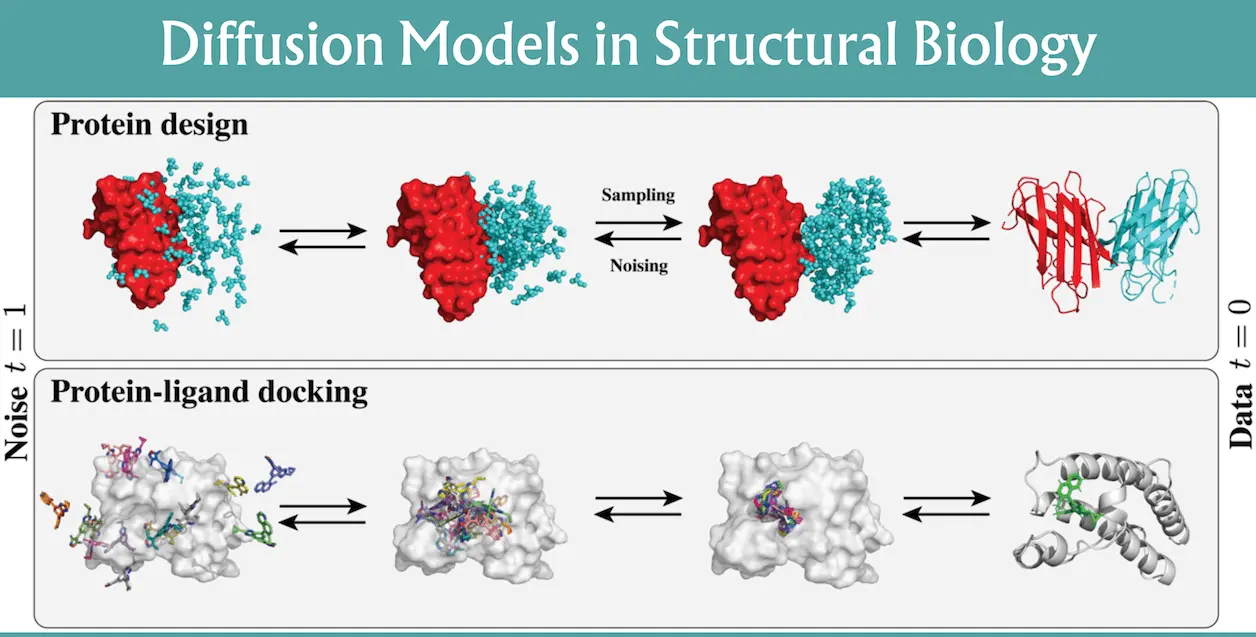
Enhancing Protein Design and Docking with Diffusion Models
Diffusion models have emerged as a significant breakthrough for protein structure prediction since they allow full proteome reconstruction via the diffusion map approach. These models are some of the most sophisticated modern protein design and docking tools, surpassing traditional approaches based on deep learning and geometric data modeling. With perfect entrainment of modes, the data-driven diffusion models help to rejuvenate computational structural biology by providing a significantly faster and synergistic way of fusing vast volumes of information for advancing science. Due to the recent advancements in the area of diffusion models, de novo protein design has not only been improved but also, in general, molecular conformer generation and blind protein-ligand docking have been significantly improved and thus are considered powerful tools in structural biology.
Diffusion Models for Structural Biology
The conformational description offered by the diffusion models has revolutionized the field of structural biology by offering prime solutions to address key problems like protein structure prediction and small molecule binding. These models are based on deep learning and geometric data modeling to reach highly automated dimensional data representation and generation. Diffusion models, therefore, present clear potentials and occasional challenges for generative techniques of the scope of computational structural biology by redressing pivotal restrictions and enhancing evaluation processes. In terms of protein structure prediction and docking, their use indicates that they are certain containers and enablers of progress in the field which include:
- Euclidean diffusion models: They form the basis of the extension of the diffusion models, particularly in structural biology-oriented mathematics. Forward and inverse operations in these models are defined using stochastic differential equations (SDEs), improving generative performance and allowing for the training of intricate distributions. Hence, these models have focused on working within Euclidean spaces and have progressed in several subfields of Computer Vision and Image processing. A clear comprehension of Euclidean diffusion models’ underlying processes and mechanisms is important when considering the quest for applications in protein structure prediction and docking.
- Geometric diffusion models: The models of geometric diffusion are discussed as a new advancement in diffusion modeling that is more suitable for structural biology. This model’s generalizations and performance are improved by incorporating geometric data types found in structure biology via symmetry encoding and Riemannian manifolds. The inclusion of geometry in descriptions of diffusive systems presents a fresh perspective in approximating molecular conformations and is likely to enhance subsequent protein folding and molecular docking estimations. It is crucial that any researcher interested in using the more intricate geometric diffusion models for computations in structural biology and related fields first grasp the principles and extensions of the given models.
Diffusion Model Approaches for Protein Backbone Generation
- ProtDiff/SMCDiff: Uses Cα atoms in Euclidean space with an Equivariant Graph Neural Network (EGNN).
- FoldingDiff: Utilizes torsion angles in the manifold of six 2D rotations (SO(2)⁶) with a Transformer network.
- ProteinSGM: Focuses on Cβ atoms in Euclidean space, refined by RefineNet.
- Chroma: Represents all backbone atoms in Euclidean space with ChromaBackbone
- DiffAb: Considers backbone atoms in the Special Euclidean group (SE(3)) using Invariant Point Attention (IPA).
- FrameDiff: Also uses backbone atoms in SE(3) with IPA and Transformer networks.
- RFdiffusion: Uses backbone atoms in SE(3) and leverages RosettaFold.
- GENIE: Represents backbone atoms in Euclidean space, combining IPA and Evoformer.
Diffusion Model Approaches for Molecular Docking
- DiffDock performs blind docking, modeling poses with 3D rotations and translations, including ligand torsion angles, treating the protein as rigid, and using the E3NN82 score network.
- NeuralPLexer, also blind, uses 3D space with contact bias for pose modeling and protein flexibility, evaluated by the IPA network.
- DPL similarly performs blind docking in 3D space for both ligand and protein, using the EGNN network for scoring.
- DynamicBind models pose with 3D rotations/translations and ligand torsion angles, accounting for protein flexibility through 3D transformations and receptor side chain torsion angles from the unbound state, also scored by E3NN.
- DiffDock-pocket/DiffBindFR is not blind, using 3D rotations/translations and ligand torsion angles for poses, with flexibility in receptor side chain torsion angles from the unbound state, evaluated by the E3NN network.
Challenges and Future Directions
Despite the remarkable achievements of the diffusion model in enhancing protein structure prediction and docking, several challenges still need to be addressed. Fine-tuning the binding poses at an atomic level and extending the developed method to different receptors and compounds are the two most important issues that need to be considered in the future. Further research can be conducted concerning flow matching, Schrodinger bridges, and stochastic interpolants to gather further improvement in the diffusion models. By enhancing the generative modeling paradigms and overcoming some of the current limitations, the diffusion models are expected to result in significant advances in computational structural biology while bringing about advancements in different aspects of computation in the discipline.
Conclusion
The diffusion models as tools applied to protein structure prediction and docking are revolutionary in providing innovative solutions to many biological puzzles. The application of these models, which are established by generative AI approaches, has had profound effects on the field of computational structural biology by augmenting the productivity in the design and development of new protein structures and by improving the efficiency of molecular docking. However, the prospects of the diffusion models are still high for future progression, as advances in the research are made on fine-tuning the modeling paradigms and elaborating the existing drawbacks. With further improvements in diffusion models under their belt, it is believed that they will remain instrumental in their use in drug design, protein design, and the study of molecular interactions, thus forming the future of structural biology.
Article Source: Reference Paper
FAQs
A diffusion model is a form of probabilistic generative model that translates a set of rough samples to a set of clear examples through a forward diffusion process. It includes using redundant noise to alter examples for their becoming noise only and using neural networks to undo this alteration stepwise. The model can take simple noise samples and map them to the target distribution with the use of probabilistic models that interpolate between the noise and data distributions.
Advantages of Using Diffusion Models in Structural Biology:
High-Dimensional Data Modeling: It also exhibits strong performances when applied to geometric data examined in structural biology, such as protein structures.
Deep Learning Strengths: By using deep learning tools, such as diffusion models, it is possible to achieve the realization of protein 3D structure generation and small molecule docking with best-in-class accuracy.
Accelerated Progress: Thus, by empowering genuine AI using diffusion models, one can fast-track advancements in computational structural biology by enabling large datasets and experiment utilization.
Follow Us!
Learn More:














{kind=link}