
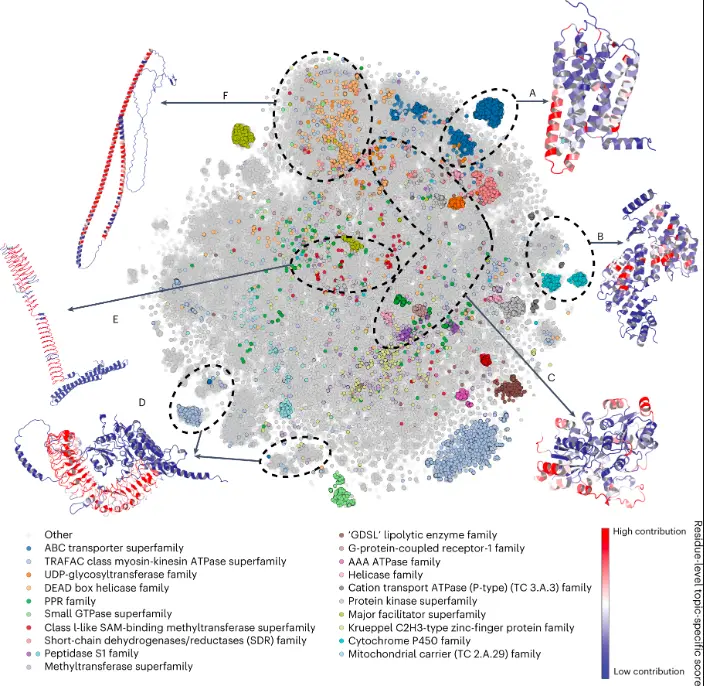
Teams of researchers across 18 institutes spread over 11 countries have worked together to assess the utility of AlphaFold2 (AF2) predictions in the analysis of distinctive structural elements, the impact of missense variants, the prediction of function and ligand binding sites, the modeling of interactions, and the modeling of experimental structural data.
A significant biological macromolecule involved in every cellular activity is the protein. It is crucial for interaction, protein function, and how missense mutations (point mutations where a single nucleotide change results in a codon that codes for a different amino acid) can affect a protein’s functionality. The primary structure of amino acids, through protein folding, forms a three-dimensional tertiary or quaternary structure. Although the experimental methods for figuring out protein structures have advanced tremendously, most of the protein universe remained unidentified. With the advent of bioinformatics and AI, such expansions of protein sequence and structural databases have become important data sources for sophisticated prediction techniques’ input and training.
DeepMind’s AlphaFold2 has been shown to predict protein structures with remarkable accuracy, similar to experimental methods. Although AlphaFold2 has been evaluated since its release by various research groups, questions remain to uncover the applications of AlphaFold 2, spanning a wide range of various structural biology problems.
Major Findings
Pedro Beltrao, an associate professor at ETH and a former group leader at EMBL-EBI, and the corresponding author of the paper titled “A structural biology community assessment of AlphaFold2 application” published in Nature Structural and Molecular Biology said that there was a lot of discussion about what the AlphaFold debut would mean for daily work of structural biologists. On social media, many people were studying the technique and their forecasts while sharing initial findings and excitement. Beltrao got in touch with a few pioneers to see if they were interested in working together on a study that would compile all the findings being circulated widely on social media.
Beltrao discovered that his concept sparked a lot of interest. It immediately gained momentum. He even received findings from social media at one point, and this work gradually developed as a result of his contacts there. It was a simple bottom-up strategy built on the preliminary findings’ anticipation. This is an excellent example of a modern approach to science. It’s like bringing everything together and utilizing everyone to the fullest while attempting to minimize duplication of work. Beltrao thinks this has a lot of value and is a fun approach to work.
Researchers have found that it is probable that the AlphaFold 2 (AF2) models are more accurate in boosting their relevance even for residues that can be described via distant homology. Here in AF2, the exact accuracy predictions are quite helpful at the residue level. Additionally, protein dysfunction is enriched for low AF2 prediction scores, suggesting that regions of doubt projections can be assumed to be disorganized
segments which are frequently used to describe areas where X-ray crystallography cannot address the problem. The AlphaFold database was initially made public with more than 300,000 modeled proteins to more than 200 million proteins with predicted structures, sampling the entire space of protein sequences and structures. AF2 made it possible to discover patterns that are quite rare such as the beta-solenoid, the shortest length of repeat beta-strand subunits.
In addition to other areas, high-confidence predictions will enable large-scale prediction of protein function from structure, identifying novel enzymes, and studying the evolution of protein structure and function. The researchers also evaluated how agreeably the AF2 predictions applied to structural biology problems, including variation effect prediction, pocket recognition, and model development using experimental data. Generally speaking, the findings from AF2-predicted structures tend to be on par with those obtained from experimental studies, consistent with the models’ high accuracy. Although AF2 provides entire protein predictions, they frequently include protein fragments positioned erratically. This uncertainty can lead to inaccurate estimations, the failure to detect structural similarity, pockets, variation effects, or inadequate model construction.
Experimental research will surely continue under AF2, however, this combination of artificial intelligence and experimental data collection is expected to be on the rise.
AF2 was utilized to predict the structure of variants, and other methods that can use the database or experimentally derived structures to predict the effects of missense mutations. An alternative strategy would be to compare or overlay the predicted structures of the reference with the mutant proteins using AF2 to assess the impact of the mutations. Nevertheless, according to some findings, AF2 does not seem to be well-equipped to forecast the structures of altered proteins. Additionally, it would have taken a lot of computational time to predict such a vast number of altered structures.
Finally, AlphaFold2 was found to perform better than traditional docking methods without even any template protein structures. The ability of AF2 techniques trained to predict unmodified intra-chain connections to predict inter-chain contacts, both for homomeric and heteromeric complexes, has also been demonstrated. So, it comes as no surprise that AF2 can be used to fold and dock heterodimeric complexes.
Final Thoughts
The impact of missense variations, functionality and ligand binding site predictions, modeling of interactions, and modeling of experimental structural data were some of the domains in which Akdel et al. examined the use of AlphaFold predictions. In conclusion, the article has discovered that AF2 models can be used to solve the burning problems in structural biology with a quality comparable to experimental models.
The improvement in AF2 by including protein complex prediction and application to unravel undiscovered structures in the protein universe will have a seismic effect on the biological sciences.
This study is of particular importance in paving the way for a new era of collaborative research with the power of social media can indeed lead to groundbreaking research in the comfort of their institutes and labs yet contribute their best research abilities. When Amelie Stein, an Assistant Professor from the University of Copenhagen, learned about Pedro’s project, was already assessing how well the modeling techniques performed on the AlphaFold structures and was eager to participate. Since the subject is developing quickly, Prof. Stein felt happy contributing to the joint project so that others might learn about and benefit from the findings.
Story Sources: Reference Paper | Reference Article
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.

Shwetha is a consulting scientific content writing intern at CBIRT. She has completed her Master’s in biotechnology at the Indian Institute of Technology, Hyderabad, with nearly two years of research experience in cellular biology and cell signaling. She is passionate about science communication, cancer biology, and everything that strikes her curiosity!





 predictions in the analysis of distinctive structural elements.){kind=link}