

The high computational cost of generating accurate atomic-level biomolecular structures remains a major challenge in AI-driven drug discovery. Researchers from Genesis Molecular AI, MIT, Carnegie Mellon University, and Imperial College London have developed DeCAF (Denoiser Cofolding All-Atom Flowmap), a framework that distills diffusion-based cofolding models into faster flow-based models capable of generating high-quality protein and protein–ligand structures in just a few steps. DeCAF outperforms Boltz-1x on benchmark datasets and matches the state-of-the-art Pearl model while using 5× fewer computational steps, paving the way for faster, more scalable biomolecular structure prediction.
Understanding biological processes such as protein folding, ligand binding, catalysis, or the development of new drugs through traditional techniques like SBDD depends entirely on the availability of precise 3D structures of the molecules at the atomic level.
This makes computational modeling of biological complexes a foundational yet the most important approach in biotech and drug discovery. But early experimental methods for SBDD, such as X-ray, crystallography, cryo-EM, and NMR, were helpful yet very expensive.
Introduction: What the Research is All About
In our earlier blogs, where we discussed RFD3, ToposBio, and PEARL, we saw that AI has already begun replacing these roadblocks by computationally predicting structures in days rather than months at the atomic level. Now we are moving towards cofolding, which refers to modeling multiple interacting molecules at once. For example, generating a protein along with a small binding ligand.
While state-of-the-art, all-atom generative models like Pearl, AlphaFold-3, and Boltz-1 have been around for a while, they never leave the ‘on paper’ stage, as most of these are still slow and require heavy computation. They have effectively proven to be a huge step towards using AI to predict biomolecular structures, but they are still impractical to be used outside of academic benchmarks and hence have become the standard.
How Model Distillation and DECAF-SEARCH Help
Current all-atom AI diffusion models like AlphaFold 3 generate structures by a gradual refinement process considering each and every atom, and the steps are judged by Neural Function Evaluation (NFEs), and each NFE takes millions to billions of computations and about 0.5 seconds. The model keeps consulting the neural networks over and over, increasing NFEs to hundreds of thousands, and this refinement is what gives the structure state-of-the-art accuracy and validity, but comes with a high computational cost and time.
But the problem appears when we want to predict millions of structures for tasks like virtual screening, computational costs become enormous to handle. Hence, the researchers come up with a model framework, DECAF (Denoiser Cofolding All-atom Flowmap Model), that distills diffusion into flow maps with only 10-20 NFEs, about 20× faster.
Model distillation in machine learning is the transfer of knowledge of a larger yet slower model, here it is Pearl (main illustrative teacher) and Botlz-1 and others (for broader applicability), into a simpler or faster model, so that one model acts as a ‘teacher’ and the learner mimics its output, but in fewer steps. This reduction in NFEs means pharma companies can actually deploy DECAF in drug discovery pipelines, overcoming the bottleneck of scaling.
Another issue that researchers highlight in the research is the inference time search algorithm designed for DECAF’s all-atom flow maps, a way to guide structure generation toward physically valid biomolecular samples, even after being computationally efficient.
“Our paper shows this Inference cost can be dramatically reduced for state-of-the-art cofolding models like Pearl without a trade-off in performance---unlocking much faster virtual screening capabilities which are critical in AI-based drug programs.”
-Dr. Joey Bose, co-author of the research and assistant professor at Imperial College, says in the Imperial College Newsletter
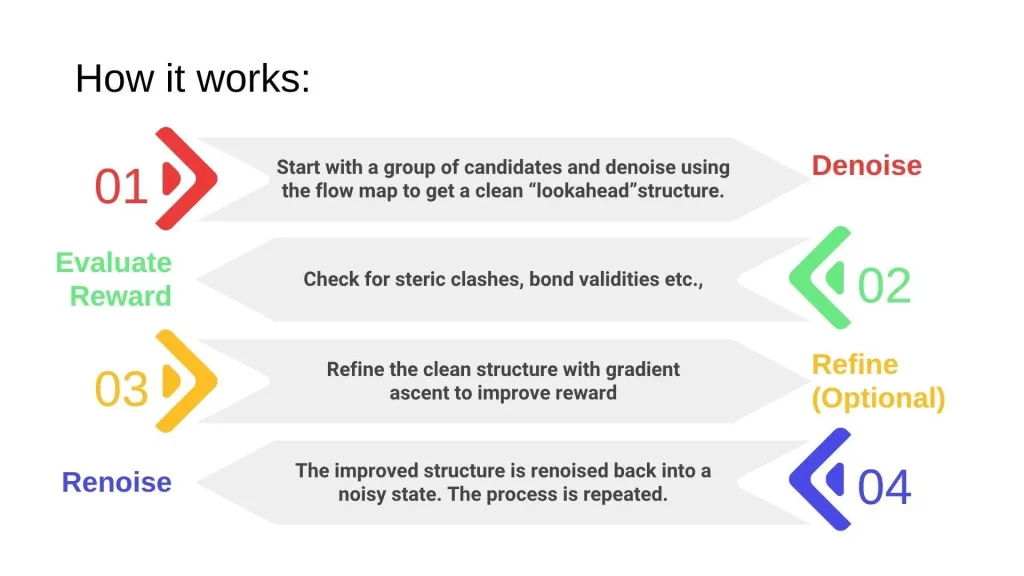
Training and Performance Parameters
Researchers mention two training innovations that made DECAF stable and effective. Firstly, reparameterization in the noise space, which helps DECAF align with the EDM-style formulation used in standard cofolding models. This makes it easier to adapt flow maps for atomic-level modeling.
Another innovation is that it uses a denoiser-based parametrization that softly forces SE(3) symmetry. This prevents the model from ‘cheating’ by memorizing the orientations. A smart solution for stable training to tackle overfitting.
Results
- DECAF-Boltz beats Botlz-1x at a low compute budget and matches full-budget performance with 20× less compute.
- On benchmarks like PoseBusters, it achieved state-of-the-art accuracy while drastically reducing inference cost.
- When applied to Pearl, DECAF-Pearl outperforms Boltz-1 and other baselines, matching Pearl’s success rate with 5× fewer NFEs.
Conclusion and Limitations:
The team created DECAF, a new model framework that takes a big, slow diffusion model, Pearl, and distills it into a flow map to generate atomic-level protein ligand pairs in just a few NFEs, about 20× faster. The DECAF-SEARCH allows us to check for physical validity much earlier and efficiently.
DECAF inherits its strengths and weaknesses from its teacher model. Here are some of the limitations mentioned by the researchers in their original research paper.
- Inherited Reward System: DECAF’s reward functions come directly from Pearl. That means any issues in Pearl’s reward design, such as the generation of non-planar sp2 bonds, are carried over to DECAF. These are not flaws of DECAF-SEARCH itself, but still matter enough to be considered.
- General σ-space denoiser: The denoiser formulation in the σ space isn’t just limited to protein cofolding, but it could potentially be extended to other biomolecular systems, such as nucleic acids or multi-chain complexes.
- Computation and success: The relationship between the inference budget and success isn’t monotonic, meaning that more compute doesn’t guarantee better results. The researchers suggest the need for smarter strategies, such as adaptive search or task-specific reward-based training.
Article Source: Reference Paper | | Reference Article | Code Avaibility: GitHub
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Saniya is a graduating Chemistry student at Amity University Mumbai with a strong interest in computational chemistry, cheminformatics, and AI/ML applications in healthcare. She aspires to pursue a career as a researcher, computational chemist, or AI/ML engineer. Through her writing, she aims to make complex scientific concepts accessible to a broad audience and support informed decision-making in healthcare.











{kind=link}