
Single-cell transcriptomics has been generating large-scale information, improving our knowledge of cellular processes across different tissues, and facilitating medication discovery, diagnosis, and prognosis. But sorting through all of this data has proven to be an enormous undertaking that frequently takes weeks or months. The vast amount of data generated—between hundreds of gigabytes and tens of terabytes—that necessitates a significant amount of time for analysis is the cause of this bottleneck. Biologists also have a challenging learning curve due to the complex set of stages involved in data processing, which needs a variety of software programs. Furthermore, because data analysis in this field is iterative, it takes a profound understanding of biology to develop pertinent questions, carry out analyses, evaluate findings, and more ideas. In order to tackle these issues, scientists from the University of California introduced Bioinformatics Copilot 1.0, a software that utilizes a broad language model. With the help of an easy-to-use natural language interface, users may analyze data without needing to be proficient in programming languages like Python or R. It is designed to work on multiple operating systems, including Windows, Linux, and Mac. Significantly, it makes local data analysis easier and guarantees compliance with strict data management guidelines governing the use of patient samples in healthcare and research facilities.
Introduction
Technologies for single-cell sequencing that rely on next-generation sequencing have improved quickly since 2009. One such single-cell omics technique that has shown great promise is spatially resolved transcriptomics; Nature Methods named it the “Method of the Year 2020.” With sub-cellular resolution, these technologies enable the visualization and quantification of thousands of transcripts, offering advantages over conventional techniques (e.g., western blot, immunohistochemical labeling) that evaluate one or a few genes in a single experiment. As a result, these tools have been applied to research in the area of brain, cancer, and embryonic development to reexamine crucial issues and provide previously unheard-of discoveries. It is projected that as more technologies hit the market, they will be used to raise the bar for biological research, diagnosis, and prognosis.
However, biomedical scientists encounter difficulties, chief among which is the magnitude of datasets, many of which are enormous and need months or even years to evaluate. While more than a thousand tools have been developed in the last ten years to support single-cell data analysis, data processing is still a major project bottleneck. Another big problem is that a lot of biologists aren’t trained in coding languages like Python or R, which makes it harder for them to analyze data efficiently.
Rise of AI
Artificial intelligence has gained significant traction over the last 20 years thanks to advancements in computing power, data storage, and the collection of large datasets. Complex machine learning algorithms have come of age, as seen by the introduction of transformer models and the emergence of ChatGPT and other similar platforms. Large Language Models (LLMs) are being used in a variety of fields, including text, voice, image, and video creation, thanks to these advancements. The effectiveness of writing, filmmaking, production, and design has all greatly increased as a result of these advancements. Researchers have started using natural language prompts to make coding work easier, especially data analysis and code completion, since big language models have become widely used. As a result, these advancements have made it possible to create Bioinformatics Copilot 1.0, a specialized application designed to speed up bioinformatics analysis by integrating LLMs.
Understanding Bioinformatics Copilot 1.0 and its Use
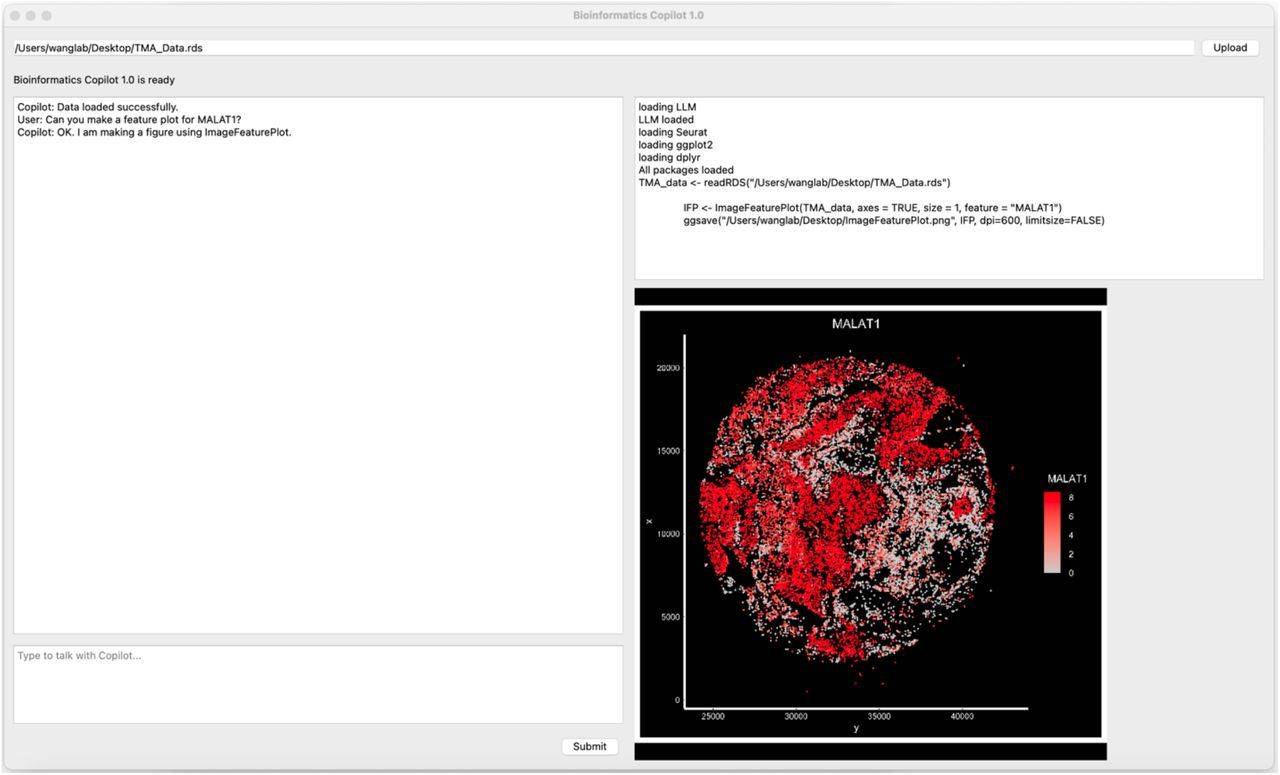
There are five main sections of the Bioinformatics Copilot user interface. There is a specific area at the top where users can upload files for analysis. Users can enter their prompts in the area located on the bottom left. The copilot will converse with users in the natural language in the upper left corner. The copilot will generate code and report progress in the upper right corner. The copilot will show the figures on the bottom right.
Gene expression levels in human cells, frequently correlated with cancer risk and therapy efficacy, are evaluated using high-throughput techniques. Scientists can assess a gene’s (like MALAT1) expression level to determine how successful a medicine or treatment is. A visualization may be made using the ImageFeaturePlot function to accomplish this.
Every kind of tissue is made up of various cell kinds, each of which has a distinct purpose. Consider tumor tissue as an example, which contains a variety of cells, including tumor cells, T cells, macrophages, and blood vessel-forming cells. To assess the condition of these tissues, it is essential to map out their cellular makeup.
Using this type of analysis, a physician can ascertain whether chimeric antigen receptor (CAR) T cells have successfully invaded the tumor or whether a treatment such as an anti-angiogenesis medication has effectively prevented the growth of new blood vessels. Users can find out the Uniform Manifold Approximation and Projection (UMAP) for a certain tissue sample in order to assess the tissue’s cell makeup. The copilot will produce a diagram that shows the various cell groupings in the tissue if you only ask for it in plain English.
Determining the exact location of individual cells within a tissue is essential to comprehending numerous biological events. For instance, one may look at the differences in gene expression between CAR T cells that are in direct contact with tumor cells and those that are not in the setting of tumor tissue. An investigation of this kind might shed light on the molecular processes underlying T-cell fatigue, cancer cell identification, and other processes. Numerous cell types, including oligodendrocytes, astrocytes, and neurons, perform various tasks in brain tissue, including signal transmission, metabolic support, and structural maintenance. Understanding the spatial distribution of cells in neurological illnesses can be essential for analyzing the onset and course of the condition and for developing focused treatments. By typing “Can you make a dimension plot?” the user can see the spatial data of the various cell types. The copilot will produce a diagram illustrating where each cell is located within the tissue.
Conclusion
The software as it stands now is limited to the analysis of Seurat objects. Larger FASTQ files, which sometimes come in sizes of several hundred gigabytes or even terabytes, are now beyond its current capability. As such, users need to obtain Seurat objects from outside service providers or bioinformatics core facilities. This version aims to streamline the frequently drawn-out communication process between bench scientists and bioinformatics specialists. Future improvements will incorporate functionality for genome mapping on a local server using user-friendly natural language to help biologists even more with data processing. Thanks to this development, biologists will be able to move forward with data analysis right after the sequencing process, which will accelerate the completion of their studies.
Furthermore, the advent of multiagent and “self-operating computers” holds promise for expanding options and enhancing software efficiency. Bioinformatics Copilot 1.0, an LLM-powered software program, has been developed by researchers to analyze transcriptome data through natural language analysis. This software is expected to expedite several research projects and contribute to advancing the biological sciences.
Article Source: Reference Paper
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}