
Transcriptional enhancers control the spatiotemporal activation of the target genes they regulate by serving as docking stations for various transcription factor combinations. It has been a long goal to Decipher the regulatory logic of an enhancer and comprehend the specifics of how spatiotemporal gene expression is encoded in an enhancer sequence. Researchers from VIB Center for AI & Computational Biology, Belgium, studied deep learning-based synthetic enhancer design. They employed a deep learning algorithm to find enhancer sequences in the fruit fly brain and succeeded in creating enhancers that selectively attacked glial and Kenyon cells. Additionally, they created enhancers that functioned in human cells. This work shows that cell-type-specific enhancers can be produced by deep learning.
Introduction
Your DNA is a large library of instructions that your body uses to build and operate. However, enhancers are required to ensure that these instructions are executed at the proper time and location. These regulatory zones attract transcription factors, proteins that serve as signposts, instructing genes to begin expressing. The role of enhancers is crucial in numerous fields, including gene therapy development and disease diagnosis.
Enhancers use sophisticated and subtle language. Because they are usually short DNA sequences, it is difficult to predict which will activate genes in specific cell types. Previously, scientists had to utilize time-consuming trial-and-error methods to uncover enhancers. This recent study used a novel method by leveraging deep learning to design synthetic enhancers from the bottom up. Using extensive datasets of recognized enhancers and cell types, the researchers trained their model. Equipped with this knowledge, the model could examine any DNA sequences and suggest modifications to transform them into useful enhancers that target particular cells.
Understanding how enhancers operate is critical for:
- Gene expression modeling and prediction
- Understanding non-coding alterations in the genome,
- Improving approaches for gene therapy
- Regulating gene expression
Conventional methods for interpreting enhancer logic:
- Mutational analysis to study individual enhancers by the insertion of mutations and observation of their effects.
- Measuring TF binding to DNA sequences in a test tube is known as “in vitro TF binding.”
- Cross-species conservation is comparing enhancer sequences from different species to find similarities.
- Reporter assays include the connection of enhancers to reporter genes and the tracking of their expression in different cell types.
- Methods of computation include developing algorithms that use sequence features to predict enhancers.
- To identify regulatory regions, genome-wide profiling analyzes enormous datasets of TF binding, histone modifications, and chromatin accessibility.
- Enhancer activation can be better understood by simulating TF-DNA interactions using thermodynamic modeling.
- The simultaneous binding of several TFs to DNA sequences is measured by high-throughput in vitro binding studies.
- Techniques for network explainability by using Predicting enhancer activity with deep learning models.
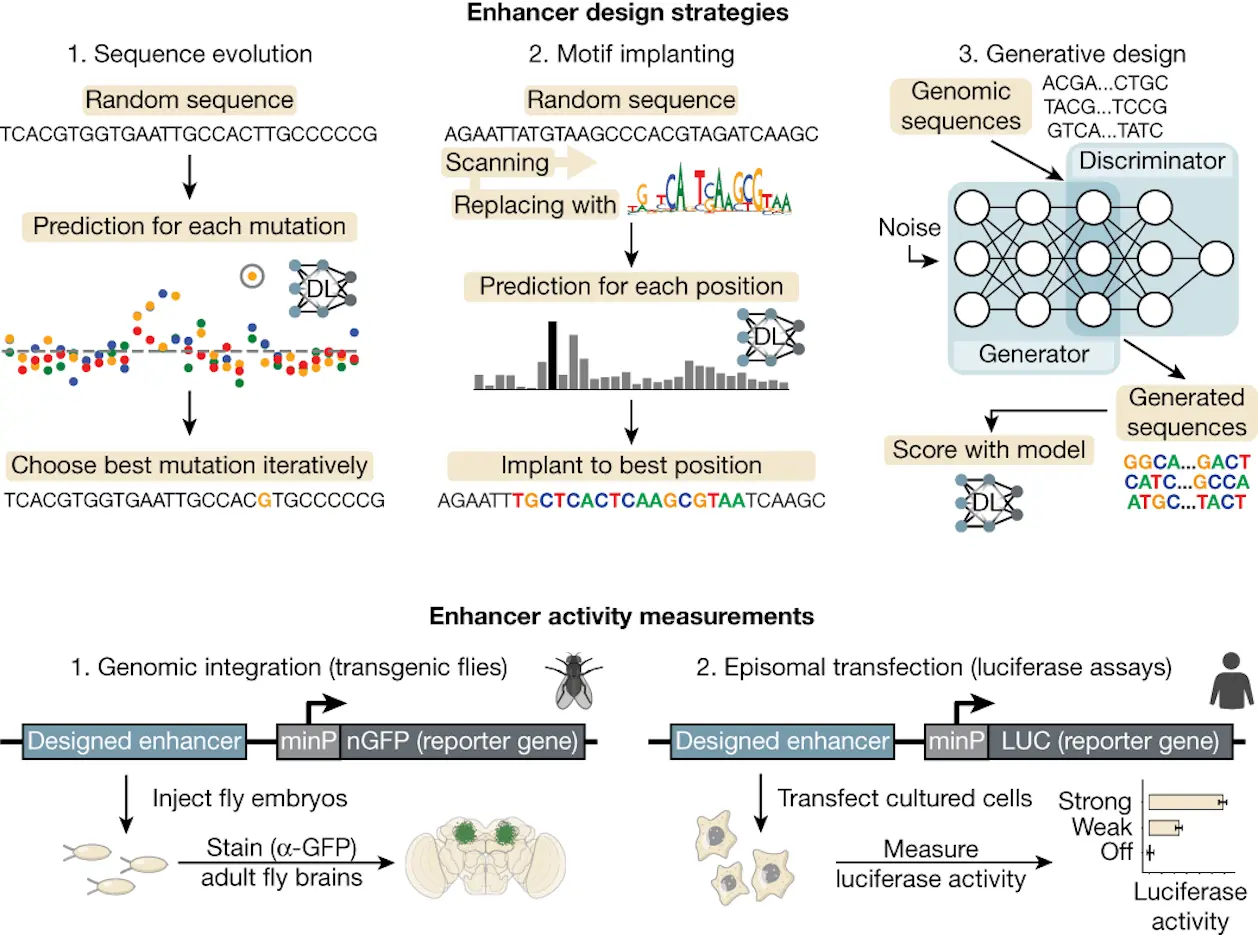
In silico evolution for designing cell-type-specific enhancers
Process of evolution:
- Saturation mutagenesis: Each nucleotide experienced a single mutation selected based on DeepFlyBrain’s KC prediction score.
- Six thousand random sequences were first generated, followed by 15 rounds of evolution.
- The scores were high for KC prediction but low for other cell types.
Examination of beginning sequences and evolutionary routes.
- The majority of random sequences had repressor binding sites, reducing KC specificity.
- The majority of alterations occurred in a 200-bp central region.
- DeepExplainer scores revealed that evolution destroys repressor sites and creates activator sites (Eyeless, Mef2, Onecut).
- Repressor sites are presumably bound by KC-specific repressors (Mamo, CAATTA5).
In vivo validation:
- In a screening of 13 synthetic enhancers, KC-specific GFP expression was seen in 10.
- Removal of repressor sites and stronger activator motifs led to an increase in activity.
- Enhancer accessibility in the genome was validated using ATAC-seq.
Adapting to different kinds of cells:
- The same random sequences underwent distinct mutations and motif modifications to become perineurial glia (PNG) enhancers.
From genomic sequences to evolution
- Researchers found regions that have little chromatin accessibility in KC but a high projected KC score.
- With just six alterations, three of the four such areas developed into positive KC enhancers.
- It was suggested that with minimal mutations, enhancers may emerge de novo in the genome.
Finding the motif:
- Repressor sites are frequently found in haphazard sequences and are eliminated throughout evolution.
- Repressor sites’ functional significance was validated by reintroducing them to interfere with enhancer activity.
Designing Cell-Type-Specific Enhancers
Various cell type codes:
- Researchers added KC-specific coding while keeping the original activity via in silico evolution, beginning with enhancers active in one cell type (T4/T5 or T1 neurons). As a result, both cell types possessed functional enhancers that were active.
- Analyzing codes On the other hand, they uncovered enhancers active in both KC and optic lobe T neurons, and they used in silico evolution to reduce T neuron coding while retaining KC activity. This led to enhancers that were particular to cell types.
Implantation of Motifs:
- The researchers chose areas with high KC prediction scores. They added strong activator motifs (Ey, Mef2, Onecut, and Sr5) into random sequences, taking into account the fact that alterations mostly affected TF binding sites.
- Ey and Mef2 showed the highest individual impacts, and their combined implantation with the right spacing increased the score even more.
- Combinations with high scores had closely spaced motifs (less than 100 bps).
- Enhancer activity was inhibited by inserting repressor motifs next to inserted activator motifs.
- It was possible to build a functional enhancer that was only 49 bp and contained the three primary activator motifs.
Human Enhancer Design with Deep Learning and Motif Implantation
DeepMEL2:
- Enhancers designed using the DeepMEL2 model produce high MEL class prediction scores.
- There is a link between predicted and in vitro luciferase reporter activity.
- It was revealed that MITF, TFAP2, and SOX10 are key activator motifs.
- In silico evolution: Iterative mutations that generate functional enhancers from random sequences.
- ZEB2 repressor motifs were introduced to reduce enhancer activity.
- In silico and in vitro testing was performed to verify the findings.
Implantation of motifs:
- The TFAP2, MITF, and SOX10 motifs were introduced into random sequences.
- Enhanced activity comparable to native enhancers was achieved.
- Only three of the implanted motifs in minimal enhancers were functional.
Extra techniques:
- The Enformer and ChromBPNet models offer complementary forecasts.
- The rehabilitated activity of a minimally mutated human “near-enhancer.”
- The GAN-based technique also created functional enhancers.
Researchers’ work represented a major advance in our knowledge of how to manipulate and control gene regulation. Even though there are still obstacles to overcome, creating cell-specific enhancers has enormous potential in many different sectors. This discovery may open the door to a new era of personalized treatment and a better understanding of the underlying workings of life, from deciphering the intricacies of diseases to creating targeted gene therapies.
Future Prospects:
- Not all boosters were developed to the fullest extent possible, and there is room for development because Developmental enhancers can be more complex than deep learning approaches realize.
- It is challenging to incorporate repressor motifs with little influence on chromatin accessibility.
- Deep learning models exhibit promises, encompassing an organism for prospective uses.
Conclusion
It was proved that the deep learning method is quite effective. The researchers developed artificial enhancers that selectively activated genes in fruit fly glial cells that support brain activity, as well as Kenyon cells that play a crucial role in learning and memory. Comprehending the transcriptional regulatory code and using this knowledge in the development of artificial enhancers has shown to be an enduring problem. Using deep learning techniques, scientists were able to successfully create artificial enhancer sequences in both humans and flies. They used a stepwise enhancer design process in conjunction with model interpretation approaches to study the pathways of in silico enhancer development in humans and Drosophila, which resulted in local optima. The selected mutations mostly eliminate potential repressor TF binding sites while producing candidate activator sites, according to nucleotide-by-nucleotide evolution. This method of evolutionary design may be a better variant of how genomic enhancers naturally arise. It was found that the human and fly genomes include ‘near-enhancers,’ which are minimally altered to function. The research went on after that. This creates intriguing new research avenues that might lead to anything from improved comprehension of human diseases to the creation of gene therapies that specifically target particular cell populations.
This discovery marks a significant advancement in our understanding of how to modify and control gene regulation. This discovery may usher in a new era of personalized therapy and a greater understanding of the basic workings of life, from decoding the complexities of diseases to developing targeted gene treatments.
Article Source: Reference Paper
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}