
The potential influence of generative models for protein design is drawing the attention of the scientific world. However, there are numerous modalities that mediate protein activity, and it is still difficult to generate multiple modalities at once. A technique for multimodal protein synthesis called PLAID (Protein Latent Induced Diffusion) has been proposed by researchers from UC Berkeley, Genentech, and Microsoft Research. It maps from a more abundant data modality (like sequencing) to a less abundant one (like crystallographic structure) by learning and sampling from the latent space of a predictor. Training for the creation of all-atom structures uses Gene Ontology and PLAID, which need both 3D structure and 1D sequence to specify side-chain atom locations.
Compared to experimental structural databases, PLAID facilitates generative model training and improves data distribution by two to four orders of magnitude by requiring only sequence inputs for latent representations. Access to additional annotations for conditioning generation is another benefit of sequence-only training. Compositional conditioning demonstrates strong structural quality and consistency on 3,617 creatures from the Tree of Life and 2,219 functions from Gene Ontology.
Introduction
Protein generative models seek to spur bioengineering innovation by putting forward designs with new capabilities. The identity, location, and biophysical characteristics of side-chain and backbone atoms are all part of the all-atom structure, which is essential for protein functions. All-atom structure formation, however, is a multimodal problem since it necessitates the simultaneous creation of sequence and structure.
Although generative modeling for protein structure has advanced significantly, there are still issues. Current techniques only produce backbone atoms and regard sequence and structure as distinct modalities. It is necessary to alternate between inverse-folding phases using external models and structure prediction when using all-atom design methods. Insilico oracle-based designability and structure-conditioning are frequently highlighted in evaluations, with little progress made in flexible controllability. Techniques that use databases of experimentally resolved structures are heavily biased in favor of crystallizable proteins. Methods that use structure as inputs could encounter difficulties when using hardware-aware neural network topologies for scalable training and inference.
PROTein Latent Induced Diffusion, or PLAID
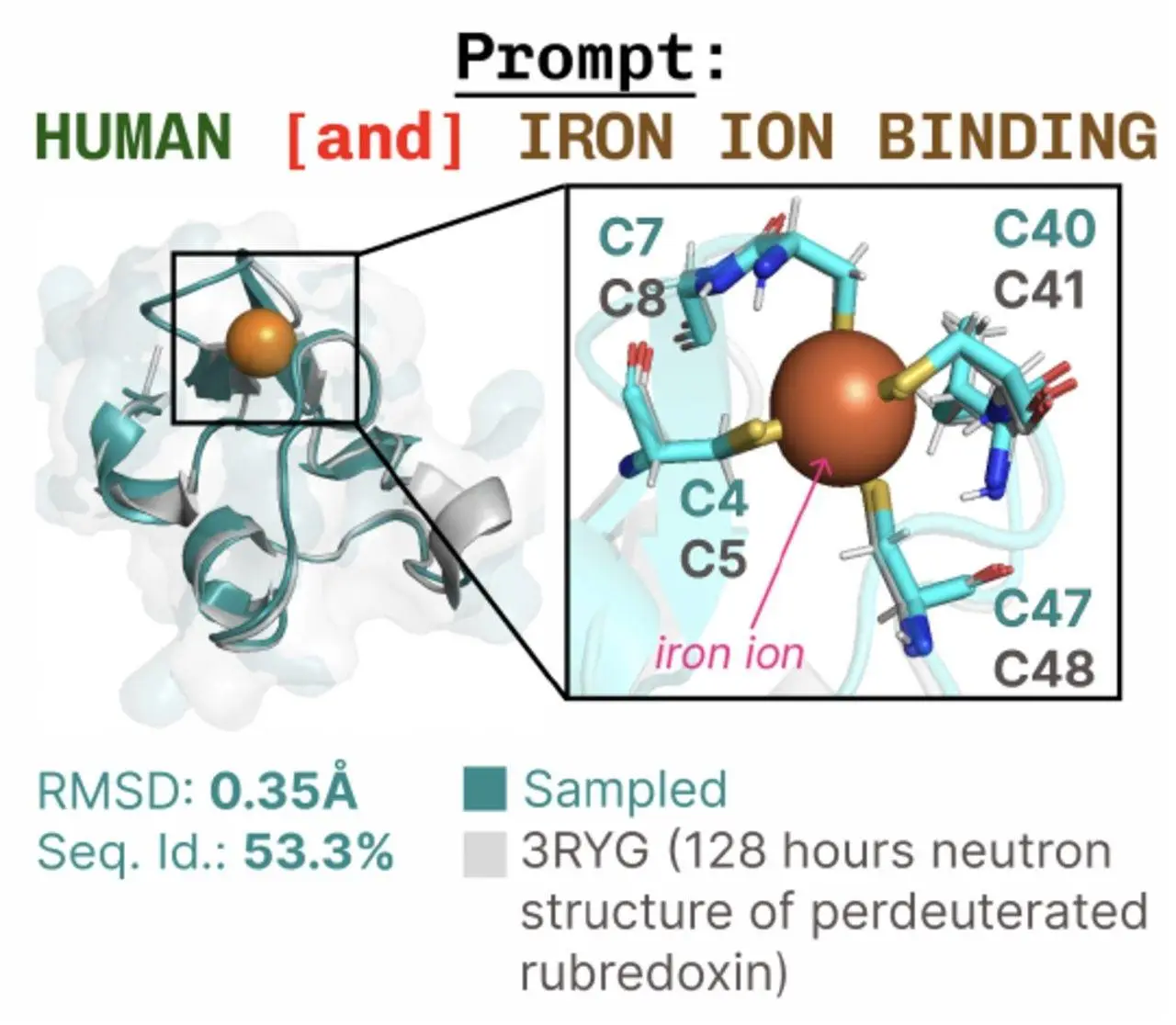
PROTein Latent Induced Diffusion, or PLAID, is introduced by researchers, which primarily shows that learning the latent space of a predictor from a more abundant input modality (like sequence) to a less abundant one (like crystal structure) can accomplish multimodal generation. Researchers specifically address ESMFold and all-atom structure formation, offering a controlled diffusion model that can generate sequence and all-atom protein structures simultaneously with just sequence inputs needed for training.
This structural protein folding model, PLAID, makes use of sequence databases to improve coverage of the viable protein space that evolution has traveled through. By using this method, the amount of experiment datasets that may be used for training is increased by two to four orders of magnitude, enabling the use of structural information that is stored in pre-trained weights. With the ability to learn side-chain orientations and sequence motifs at active sites, PLAID can unconditionally produce a wide variety of high-quality samples. It is flexible enough to accommodate growing sequence datasets and exhibits motif scaffolding. This approach makes use of enhanced inference and training infrastructure for transformer-based models as well as increasingly multimodal protein folding models, such as those involving molecular ligand binding and nucleic acids.
Performance of PLAID
Regarding performance, PLAID, a model for generating protein structures, has been contrasted with Multiflow and ProteinGenerator. Because Multiflow does not generate side chain positions, PLAID’s performance diminishes as protein length increases. Training on longer sequences is made easier by PLAID’s adaptable Diffusion Transformer design and longitudinal downscaling.
More big protein samples are included in the enlarged collection, which strikes a balance between diversity and quality. The diversity of secondary structures in PLAID is more in line with the profile of real proteins. This suggests that samples with a high β-sheet content are frequently difficult for current protein structure generation models to generate.
The structural similarities between PLAID and natural proteins serve as the foundation for PLAID, a revolutionary approach to predicting expressibility in the real world. These similarities are measured by the backbone TMScore, where higher uniqueness is indicated by lower values. Low-quality samples that fictitiously score highly on novelty measures, however, might have an impact on this. Perhaps as a result of biases being eliminated from training data, PLAID’s biophysical characteristics are more similar to those of natural proteins.
The significance of comprehending and resolving structural similarities in PLAID is shown by the strong correlation between real-world expressibility and distributional conformity, another axis of real-world expressibility.
Limitation
The performance of the prediction model PLAID is limited, especially when frozen decoders are created. However, model performance can be enhanced with explicit fine-tuning for latent generation. Alternative conformations are not sampled by the deterministic structural decoder currently in use. A decoder that returns a distribution of structural conformations might be the answer; this could be improved or developed with field progress. The hierarchical structure of the Gene Ontology vocabulary and the existence of several pertinent GO concepts in a protein are not taken into account by the GO term one-hot encoding. This problem might be resolved with a multi-class conditioning technique. Potential drawbacks in practical use situations can be addressed by separating the classifier-free guiding scale for organism and function circumstances.
Conclusion
PLAID is a unique method for diffusing proteins in the latent space of a prediction model in a multi-modal, controlled manner. Advances in sequence-to-structure prediction capabilities, model scalability, and data availability are all utilized in this approach. GO keywords are used as a stand-in for many language annotations in the implementation, along with quick attention kernels for transformer-based architectures. PLAID may be used to predict complexes from sequence and can be used in any prediction model. Traditional in-painting jobs can also be used, but the strategy focuses on allowing new capabilities utilizing Pfam and GO words as proof-of-concept.
Article Source: Reference Paper | Model weights and code are publicly available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}