
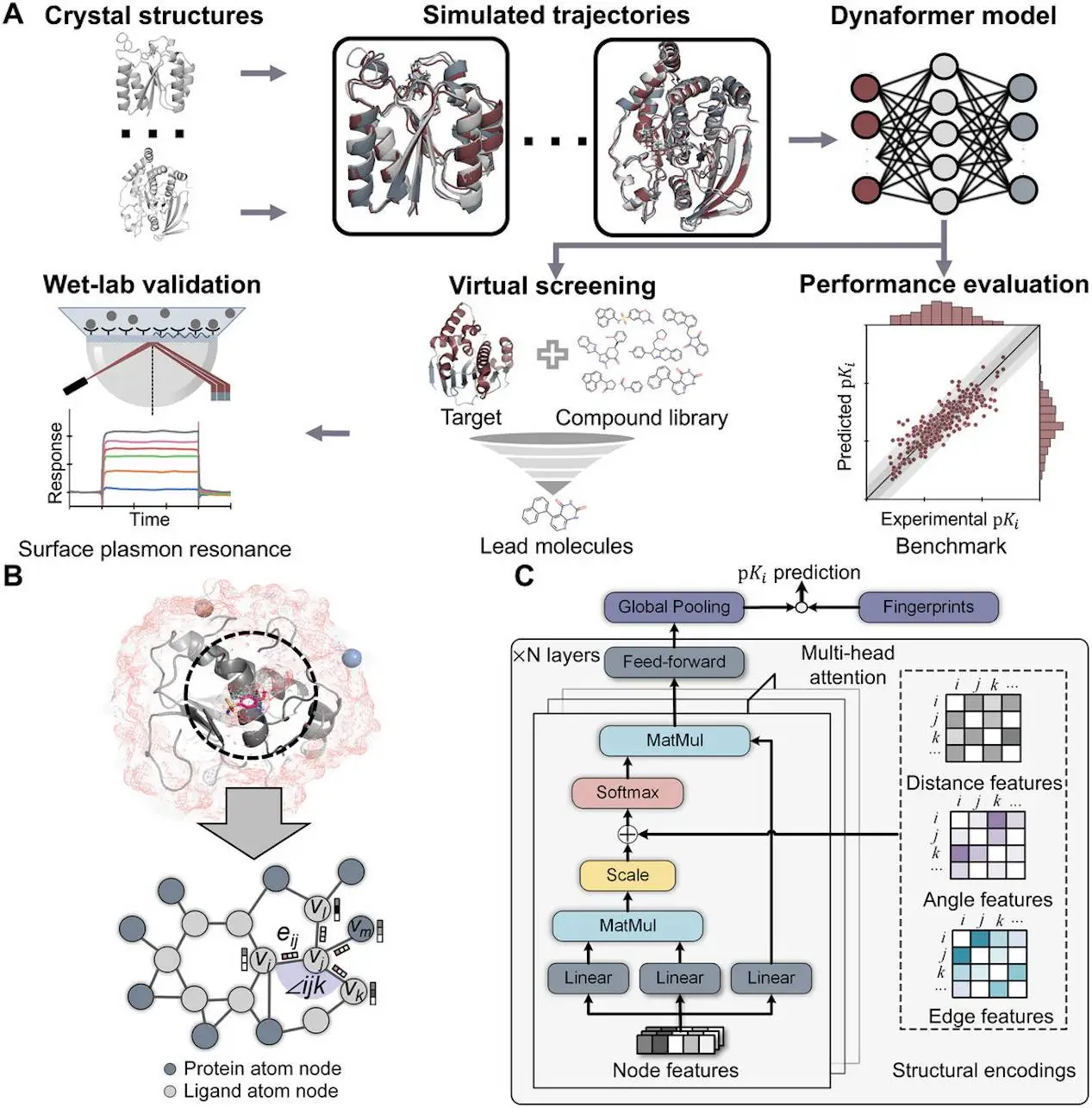
One major obstacle in structure-based drug design is accurately predicting the affinities of protein-ligand interaction. The accuracy of data-driven affinity prediction methods has improved recently, but its limitations remain. This is partly because these methods rely solely on static crystal structures, whereas thermodynamic ensembles between proteins and ligands typically dictate the real binding affinities. Using molecular dynamics (MD) simulation is one efficient method to estimate such a thermodynamic ensemble. Here, the researchers from Westlake University and collaborators have curated an MD dataset with 3,218 distinct protein-ligand complexes. Based on the MD trajectories, they have further developed a graph-based deep learning model called Dynaformer, which is capable of predicting binding affinities by learning the geometric properties of the protein-ligand interactions.
Introduction
In many biological processes, including signaling cascades, enzyme catalysis, and cell shape maintenance, protein-ligand interaction is essential. It is crucial to comprehend the binding processes of ligand molecules to create new medications with a high affinity and selectivity. For this reason, one of the most important jobs in drug development is creating precise computational tools for modeling protein-ligand binding and estimating the matching affinities.
Recent developments in computational techniques have made it possible to estimate the affinity of protein-ligand binding using knowledge- and physics-based methodologies. Coarse-grained physical models are used by molecular docking algorithms to forecast and assess the binding conformation of tiny molecules. Nevertheless, these techniques typically involve specialized knowledge and manually adjusted parameters, which frequently produce skewed outcomes. MD, or all-atom molecular dynamics, is another well-liked computer method for examining the dynamical and structural characteristics of biological systems. The primary method for revealing all of the atomic details at a fine spatial-temporal resolution is Newtonian mechanics. This is the foundation for many binding affinity calculation techniques, including MM/PB(GB)SA and alchemical free energy. However, the application of molecular dynamics simulation to high-throughput virtual screening is hindered by its typical high computational resource consumption.
Learning about Data-Driven Approaches
The complicated process of protein-ligand interaction necessitates data-driven methods to estimate binding affinity. These approaches, which have become more accurate by directly learning binding modes from 3D structures, are not very generalizable and frequently call for complex feature engineering. Existing approaches, like feature-based, voxel-based, graph-based, and docking-based approaches, ignore dynamic patterns in favor of fine-tuning models on X-ray crystallographic structures. Protein-ligand binding affinity depends on a thermodynamic ensemble or a conformation distribution in an equilibrium state. Unfortunately, the lack of processing resources makes large-scale MD trajectory datasets scarce. Using machine learning, the molecular dynamics simulation framework TorchMD improves empirical force fields and forecasts conformations in the target protein’s flexible regions. Many deep learning-based techniques have been investigated to learn MD trajectory features and extend trajectories beyond MD-accessible time spans. These methods offer viable ways to build extensive MD simulation datasets using classical or machine learning force fields, as well as get beyond the computing constraints of MD simulations. Nevertheless, thermodynamic ensembles have not been considered by many data-driven techniques that are now in use to predict the affinities of protein-ligand interaction.
Key Highlights of the Work
- Researchers thus investigate how combining MD-simulated thermodynamic ensembles with a data-driven deep-learning model may enhance the prediction of protein-ligand binding affinity.
- First, researchers assembled a massive MD trajectory dataset based on the PDBBind dataset, which included 3,218 distinct protein-ligand complexes.
- Then, using this MD trajectory dataset as training data, researchers presented the Dynaformer graph transformer system.
Understanding Dynaformer
To capture the binding modes between ligands and proteins, Dynaformer uses a roto-translation invariant feature encoding scheme that accounts for a variety of interaction characteristics, such as angles between bonds, interatomic distances, and different kinds of covalent or non-covalent interactions. On the CASF-2016 benchmark dataset, Dynaformer demonstrated better scoring and ranking power than other approaches currently in use. Scientists
also covered the underlying thermodynamic binding mechanisms that might be responsible for the performance boost that Dynaformer has achieved through a number of case studies. Additionally, researchers conducted experimental validations of multiple novel hits against heat shock protein 90 (HSP90), a promising target for cancer drugs, found using Dynaformer. Researchers have shown that the model is a useful tool to speed up the drug discovery process and can be used for a real-world hit-finding challenge.
Outcomes of the Work
- Preliminary in silico tests revealed that the model surpasses previously published approaches by displaying cutting-edge scoring and ranking capabilities on the CASF-2016 benchmark dataset.
- In addition, twenty candidates are found, and their binding affinities are further experimentally confirmed in a simulated screening conducted using Dynaformer on heat shock protein 90 (HSP90).
- Twelve hit compounds, including several novel scaffolds, were found using Dynaformer’s virtual drug screening. Of these, two are in the submicromolar range. The results look promising.
- Collectively, these findings showed that the strategy presents a viable means of expediting the early stages of the drug discovery process.
Conclusion
The basic issue of predicting the affinity of protein-ligand interaction in the early stages of drug discovery is addressed by the graph transformer model called Dynaformer. A comprehensive MD trajectory dataset of 3,218 complexes containing proteins and bound small molecules is used by the model to learn the underlying physico-chemical patterns of binding. The model demonstrated a considerable improvement over baseline techniques on the CASF-2016 benchmark dataset, underscoring the value of using MD trajectory data to improve binding affinity prediction’s scoring and ranking power. It was shown that Dynaformer was effective in finding hit compounds with advantageous binding affinities and unique scaffolds in the real world when used against HSP90. The research highlights the potential benefits of using deep learning models in conjunction with MD trajectories to improve binding affinity prediction. Researchers have demonstrated the power of Dynaformer as a scoring function and its capacity to identify new hit compounds in both a real-world drug discovery setting and a benchmark dataset. Researchers believe that by adding more high-quality data and properly crafting training problems, prediction performance can be further enhanced.
Article Source: Reference Article
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty
-
Deotima Chakraborty











{kind=link}