
Reduced representation methylation sequencing for CpG islands or imprinted areas is made possible by Oxford Nanopore sequencing, which offers a distinct advantage over other techniques by identifying DNA methylations from the ionic current signal of individual molecules. Here, scientists present DeepMod2, a thorough deep-learning framework that uses the ionic current signal from Nanopore sequencing to detect methylation. A Transformer model and a BiLSTM model are used by Oxford Nanopore Technologies’ DeepMod2 program to analyze POD5 and FAST5 signal files. Using model pruning and phased scans to infer methylation calls or epihaplotypes, it operates efficiently on a CPU. DeepMod2, despite being closed-source, performs better than cutting-edge techniques like Guppy and Dorado, indicating its potential for use in cellular signal analysis.
Introduction
DNA methylation is an essential process that involves the addition of methyl groups to particular DNA nucleotides. This process has an impact on several biological processes, including cancer, aging, genomic imprinting, genome stability, and repression of transposable elements. 5-methylcytosine (5mC) is the most common type of DNA methylation in humans. It is a characteristic of cancer and frequently results in localized hypermethylation of tumor suppressor genes. Today, the US Food and Drug Administration (FDA) is testing or has approved a number of medications that target DNA methylation regions as a potential therapeutic target for cancer. One of the first methylation inhibitors utilized in cancer clinical trials, for instance, was 5-Aza-2′-deoxycytidine, which paved the way for exon skipping and isoform switching. Methylation is a possible biomarker for cancer cells since it can differentiate them from normal cells through both global and local 5mC hypomethylation and hypermethylation. Due to a lack of trustworthy high-throughput techniques and reference data sets, N4-methylcytosine (4mC), 5-hydroxymethylcytosine (5hmC), and N6-methyladenine (6mA) are less researched than 5mC while playing crucial roles in controlling gene expression.
Although they have significant drawbacks, methylation microarrays, and short-read sequencing have been utilized to profile 5mC in CpG sites at single base resolution. These techniques have limits when it comes to testing repetitive areas and epihaplotypes, call for specific DNA preparation, and are susceptible to PCR biases and conversion efficiency issues. These restrictions can be overcome by long-read sequencing technologies like Oxford Nanopore Technologies (ONT), which differentiate between methylation and unmethylated cytosines using an ionic current signal. In a variety of genomic contexts, a recent survey revealed strong agreement between bisulfite sequencing and ONT methylation prediction.
Adaptive sampling is used in nanopore sequencing to pick DNA molecules according to the read sequence, enabling real-time acceptance or rejection of molecules in less than a second. Reduced representation methylation sequencing (RRMS), which is comparable to reduced representation bisulfite sequencing (RRBS), can be achieved by designing target genomic areas such as CpG islands or CpG-rich promoters. Targeting 310 Mbp, or 10% of the human genome, which includes all CpG islands, CpG shelves, CpG coasts, and several promoter regions, Nanopore published sophisticated techniques for adaptive sequencing on the human genome in 2022.
Disadvantages of Peer-Reviewed and Open-Source Methylation Detection Tools
- Peer-reviewed, open-source methylation detection techniques have fallen far behind the technological advancements in ONT sequencing over the last few years. For instance, only R9.4 flowcell models are provided by Nanopolish, DeepMP, DeepSignal, DeepMod, Tombo, methBERT, and Rockfish; ONT ended support for these flowcells in 2023.
- The signal profile of the most recent R10.4 flowcells is entirely different from that of the earlier models, and they have a longer protein pore with two pinch sites for two signal measurements.
- Furthermore, no open-source program is able to analyze BAM move tables or POD5 files, which ONT basecallers use to store basecall and signal data instead of FAST5 files.
Understanding DeepMod2
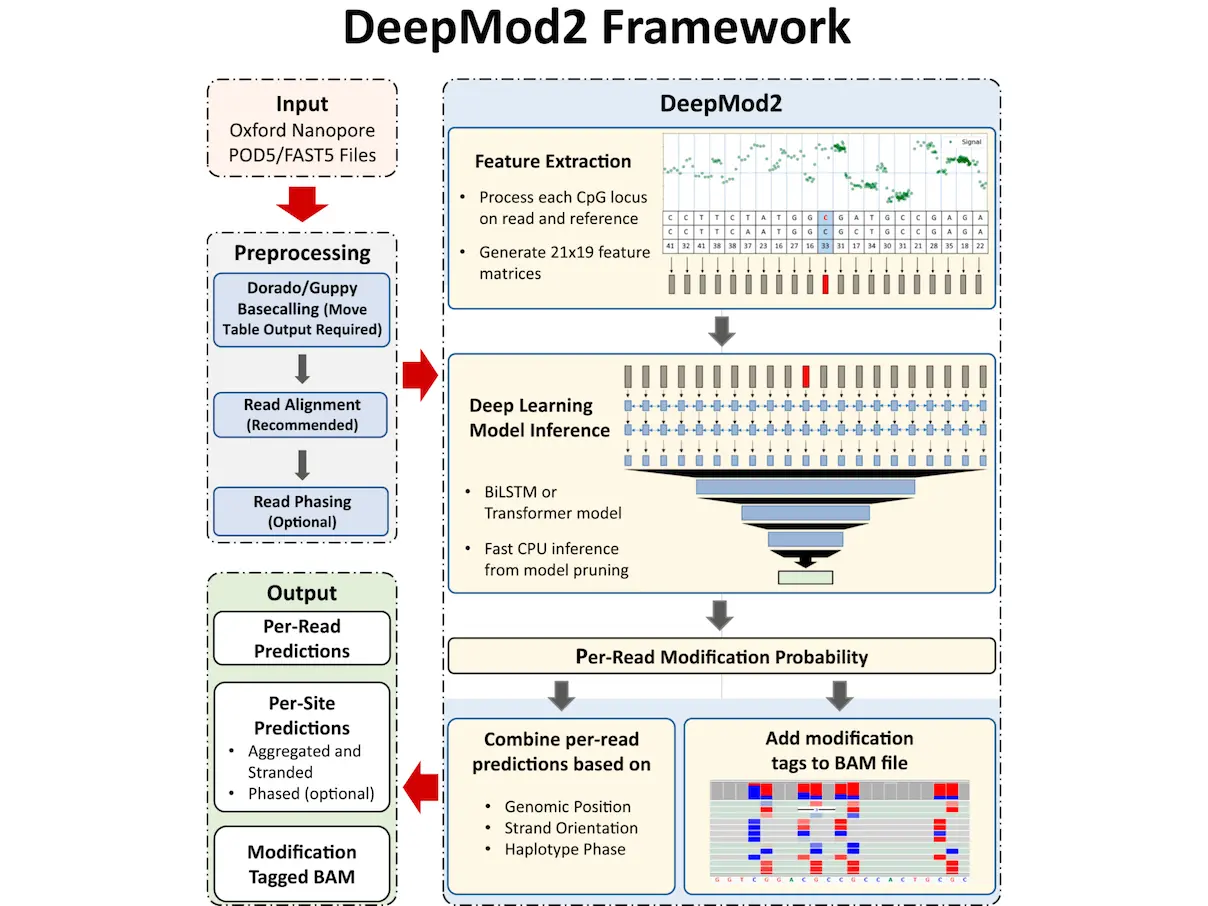
DeepMod2 is a comprehensive deep learning framework for methylation detection from Oxford Nanopore sequencing. Compared to its predecessor, DeepMod, Oxford Nanopore Technologies’ DeepMod2 tool represents a major improvement. Oxford Nanopore flowcells of different kinds and signal data formats can be analyzed using it. The instrument is evaluated using RRMS of HG002, a commonly used human reference cell line, and whole-genome Nanopore sequencing of the NIH3T3 cell line. DeepMod2 performs on par with Oxford Nanopore Technologies’ Guppy and Dorado, which are currently their state-of-the-art techniques. On human cell lines, DeepMod2 achieves between 95% and 99% F1-score, and there is a strong association between reduced representation and whole-genome Nanopore sequencing of HG002. Additionally, a strong association has been shown between reduced representation and whole-genome Nanopore sequencing of HG002, indicating that this approach may be a practical means of doing affordable, large-scale methylation profiling of intricate genomic areas.
Advantages of DeepMod2
When compared to other open-source ONT methylation callers like Nanopolish, Rockfish, f5c, DeepSignal, and methBERT, DeepMod2 with Guppy and Dorado has a number of advantages. For example, DeepMod2 may retain the methylation information of every CG motif on a read in the MM and ML tags of its BAM output. Fast searching into any genomic site is made possible by sorted and indexed BAM files, which also make it easy to analyze methylation allele-specifically and visually validate methylation using genome browsers like IGV. In contrast, some open-source methods only generate a plain-text per-read output that makes it difficult to assess the methylation of different reads from the same region because it generally has several hundred million lines of unordered predictions. More crucially, both DeepMod2 and Guppy/Dorado can detect methylation from unmapped reads or unaligned segments of reads and preserve this methylation information in the BAM file. The runtime study demonstrates that DeepMod2 and Guppy/Dorado can accurately call 5mC methylation without a reference genome or matched data.
Accurate alignment of methylation-tagged BAM files to multiple reference genomes can be achieved without requiring a repeat of methylation detection for every reference genome. This can be achieved because, during alignment, methylation tags stay unaltered and annotate 5mC with respect to read coordinates. When the sole reference sequence available is from a closely similar species or when the reference genome for a particular species has significant gaps or is erroneous, this can be quite useful. Moreover, read alignments that are incomplete or imprecise are sometimes caused by the presence of structural variations and chromosomal rearrangements, and some methylation patterns might only show up after aligning to a modified or distinct reference genome.
DeepMod2 performs better in terms of methylation detection models than proprietary programs like Guppy and Dorado. Guppy and Dorado save a significant amount of time by combining basecalling and methylation calling into a single phase, removing the requirement to reopen signal files and process signals for methylation. However, Dorado/Guppy causes significant computational redundancy because it necessitates re-basecalling the data from scratch for various forms of methylation or synthetic DNA modification. DeepMod2 shows that even on CPUs, methylation detection is a simpler work that can be completed rapidly and effectively.
For the basecaller model, Dorado v0.3.4 currently offers four different methylation detection models: “5mCG_5hmCG,” “5mC_5hmC,” “5mC,” and “6mA.” Running more than one of these models would need basecalling the same reads twice because they are unique and maybe non-interchangeable. In about 6 hours with a GPU and 13 hours with CPUs, DeepMod2 performs methylation detection that is isolated from basecalling and reuses basecalls with the aid of move tables.
Conclusion
Using ionic current signals in genomes, Oxford Nanopore’s DeepMod2 technique provides reliable 5mC modification detection. With signal alignment data from Guppy or Dorado basecallers, it can parse POD5 and FAST5 files. R10.4 and R9.4 series flowcell models are provided by DeepMod2, which also generates comprehensive per-read and per-site forecasts. It performs better than other methylation identification techniques, such as Rockfish, Guppy, and Dorado, which frequently perform within 0.2% of one another in terms of per-site F1-scores. It is possible to successfully apply DeepMod2 models to other species that were trained on human genomes. In the genomes of HG002, HG003, and HG004, putative imprinted areas are also found, showing significant overlap with previously identified imprinting control regions (ICRs). In general, DeepMod2 presents a viable approach for methylation identification within genomes.
Article source: Reference Paper | The DeepMod2 software is available on GitHub and is distributed under the MIT License.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}