
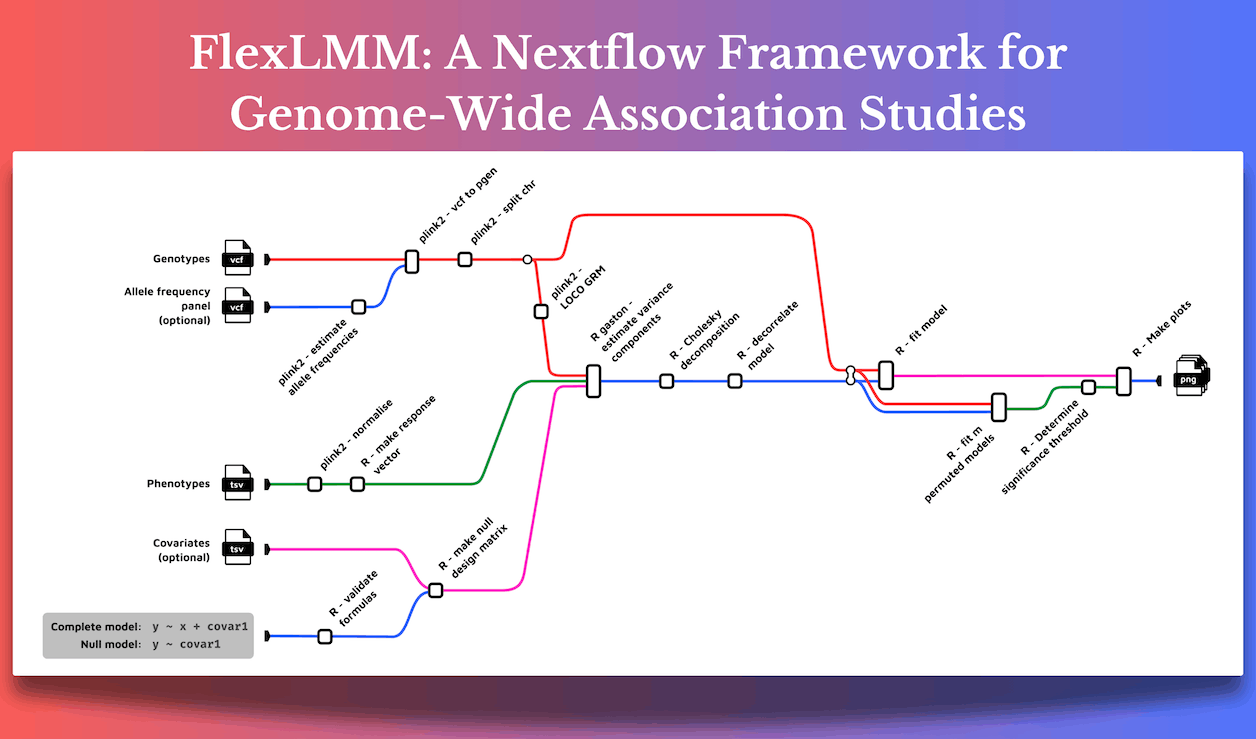
Genome-wide association Studies (GWAS) consider sequencing the DNA of a person and then comparing it with a population to find genetic markers associated with a disease or trait, and this is why they have become an integral part of modern genomics. However, the ongoing development of GWAS has led to the growth of problems associated with it. Among the major issues is the issue of population structure: the subtle genetic differences between groups can lead to false conclusions if not properly handled. Enter FlexLMM, a flexible framework built by researchers of the European Bioinformatics Institute, Cambridge, on the Nextflow platform, designed specifically to do this with Linear Mixed Models (LMMs).
GWAS: Finding the Genetic Needle in a Haystack
Imagine you’re a detective trying to find the cause of some mysterious illness. You have a gigantic database of people’s genetic information, and you know that the cause of the illness is somewhere hidden in their DNA. This is precisely what scientists do in GWAS: they glance over genetic data from thousands of people to find variations, called single nucleotide polymorphisms (SNPs), associated with diseases or traits. The idea is to find a genetic “needle in the haystack.”
However, it poses a difficulty when individuals in your database aren’t entirely independent of each other. For example, maybe they have common ancestry or are related to each other. This introduces confounding variables. In our analogy, it’s like trying to determine what is causing the illness while knowing certain families or communities in your database tend to share similar environments or lifestyle habits. For instance, these similarities might make you think that a certain variant predisposes you to the disease when it’s actually really common in that family or population.
That is where FlexLMM helps. Using LMMs, including the common backgrounds of the individuals, makes it easier to detect real effects rather than getting derailed by the population structure.
LMMs: Accounting for Relatedness
To extend the analogy, imagine that you’re not only studying the people but also the trees that their relatives have. Some of your subjects are closer relatives, almost cousins or siblings, while others are more distant. Such relationships may affect your findings. A particular disease seems to manifest in a family not because of genetics in one individual but through shared genetic and environmental inputs throughout the whole family.
LMMs account for these relationships. It does so by partitioning the variation in your data into two parts:
Fixed effects are the main factors you are investigating (for instance, a given SNP that may be responsible for causing the disease).
Random effects – that control for any underlying structure, such as the degree of relatedness among individuals. A way to phrase it is, “I am looking at this SNP, but I am also going to control because these people are of the same ancestry.”
This prevents false signals because some of the traits, such as those through genetics, are more common in a specific subgroup of individuals. It uses LMMs in order not to produce false positives and to ensure that all associations it finds will be stable and reliable.
FlexLMM: Bringing Flexibility into GWAS
FlexLMM is basically about flexibility. It’s based on Nextflow, a workflow management tool specially devised for the running of computational pipelines in biology. Think of Nextflow as the large, complicated kitchen. In this kitchen, dozens of chefs are instructed on different tasks: chopping onions, preparing sauces, and cooking the main dish. Nextflow ensures that all these tasks are appropriately coordinated and done properly, and nothing is burnt in the process.
With the management capabilities of Nextflow, FlexLMM scales up to efficiently process huge datasets for GWAS. This is important because most contemporary GWAS involve millions of SNPs and thousands of participants and are, therefore, computationally expensive. It is the guarantee that Nextflow gives to FlexLMM that, therefore, allows researchers to focus more on the science rather than getting bogged down by the technical complexity of data management.
Hence, flexible models can be defined as fitting best to the needs they have by using FlexLMM. In fact, it doesn’t put researchers into a mold of applying a predetermined method but instead puts them in a position of giving them tools to modify their analyses. For example, if a researcher really wants to test a new hypothesis related to the interaction that exists between genetic variations and environmental factors, it’s easy with FlexLMM.
Why FlexLMM Matters
FlexLMM provides the correct answer, regardless of population structure or relatedness. As an illustration, suppose a company is interested in determining which of its product features accounts for higher sales of the products in various regions. Without adjustment for the fact that people in certain regions have similar buying behavior, it would misattribute to the preferred feature in the region, which is those higher sales. Perhaps this represents a regional rather than the characteristic itself. Similarly, FlexLMM prevents making spurious inferences from genetics data by including the “regions” (population structure) in analyses.
It especially comes to light in the application process for potential applications. The FlexLMM can be applied not only in disease research but in various studies that exploit traits such as height, intellectual ability, or reaction to medication. Personalized medicine will eventually dominate the treatments developed and based on the genetic background of individuals; therefore, tools like FlexLMM to properly identify genetic association will prove to be an inevitable part of that process.
Conclusion
The development and application of FlexLMM bring out new dimensions in the context of flexibility and scalability in modern genomic research. Larger and even more complex datasets will fail rigid tools that cannot adapt to different study designs or models. FlexLMM teaches that it is essential to tailor workflows and scale computations over diverse environments, from those equipped with more modest local machines to better-equipped big clouds, to make truly groundbreaking discoveries.
In conclusion, FlexLMM is no new tool in the GWAS toolkit; rather, it is a shift toward more flexible, scalable, and accurate ways of analysis.
Article Source: Reference Paper | Source code and documentation for the FlexLMM are available on GitHub.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Neermita Bhattacharya is a consulting Scientific Content Writing Intern at CBIRT. She is pursuing B.Tech in computer science from IIT Jodhpur. She has a niche interest in the amalgamation of biological concepts and computer science and wishes to pursue higher studies in related fields. She has quite a bunch of hobbies- swimming, dancing ballet, playing the violin, guitar, ukulele, singing, drawing and painting, reading novels, playing indie videogames and writing short stories. She is excited to delve deeper into the fields of bioinformatics, genetics and computational biology and possibly help the world through research!











{kind=link}