
Data-driven drug discovery provides an effective path for drug development using large bioassay datasets containing the bioactivity profiles of millions of compounds. Recently, researchers have developed BEAR (Bioactive compound Enrichment by Assay Repositioning), a novel method of finding potential drug candidates by exploiting those bioassay datasets. BEAR can discover a diverse range of potential dr
ugs and is good at finding drugs that work on different types of proteins without consulting any structural information. The authors tested BEAR’s performance on many different proteins and found that it worked better than other machine-learning models. The candidates identified by BEAR were also experimentally validated, highlighting its great potential as a tool for ligand-based drug discovery.
Challenges of Early Drug Discovery and Virtual Screening
The process of identifying molecules with the potential to become effective, non-toxic drugs is known as early drug discovery. This is a complex task and is a considerable challenge. This is because it conventionally involves screening hit compounds from large compound libraries, which are enormously time-consuming and expensive. The traditional high-throughput screening techniques, which directly measure the bioactivity of up to millions of compounds, have a very low hit rate, typically under 1%. This suggests that even massive biopharmaceutical companies may screen extensive compound libraries but only cover a small fraction of the compound space. Drug developers now generally combine virtual screening (VS) and experimental validation in drug development programs to address this challenge.
The VS approach involves predicting how well compounds bind to specific target proteins by analyzing their physical and chemical properties. Docking is a VS approach that uses known molecular interaction principles to predict binding affinities. Another VS approach is ligand-based virtual screening (LBVS or LBDD) which assumes that compounds with similar structures and properties would bind to similar targets. LBDD techniques do not require 3D structures of targets, making them suitable for targets with unresolved structures like membrane proteins. In fact, given that more chemogenetic data is becoming available, LBVS/LBDD is becoming an attractive technique. Among LBVS methods, the quantitative structure-activity relationship (QSAR) model is broadly used because of its speed (high throughput), good hit rate, and ability to derive novel compounds. However, QSAR models are limited to structurally similar compounds with related scaffolds correlating well with the desired biological properties.
BEAR: A Novel LBVS Algorithm
Furthermore, proteochemometric modeling is also a promising approach as it uses descriptors for both targets and ligands and data-driven deep learning models like GENTRL, which have shown high performance in VS. However, the issue with deep learning models is that they require numerous parameters compared to the training data size and are more vulnerable to overfitting or memory problems. Moreover, using in vitro and in vivo assays to measure the bioactivity of compounds is popular in early drug screening, with bioassay datasets being generated rapidly. However, the large volume of these extensive bioassay datasets has not been used in data-driven drug discovery which is because of the heterogeneity of the raw data and the sparsity of the NxM data matrix. These are significant limitations.
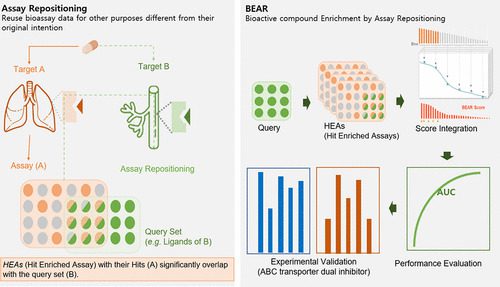
Recently, a paper published in the Journal of Chemical Information and Modeling reports a novel algorithm called Bioactive Compound Enrichment by Assay Repositioning for VS in drug discovery. BEAR addresses the issue of the heterogenous, sparse, and irregular nature of bioassay data sets, and unlike other existing algorithms, which need physicochemical properties of compounds, BEAR only needs bioactivity datasets and compound identifiers.
This BEAR approach in drug discovery essentially involves using bioassay datasets to screen for potential drugs by searching for targets other than their original intended use. This is known as assay repositioning and allows searching for bioassays with hits that significantly overlap with a query set of ligands or query compounds set as input. These bioassays with significant hits are known as Hit-Enriched Assays (HEAs) and are believed to capture the physicochemical properties and/or bioactivities shared among the query compounds. By integrating the bioactivity information of all such HEAs, BEAR can predict candidate compounds with bioactivities similar to the query set.
The BEAR algorithm consists of five steps;
- Takes a list of input compounds as the query set
- Selects bioassays, that is, HEAs, with a Hit Enrichment Score above a cutoff threshold.
- Arranges the compounds of each HEA by their activity levels and binned by equal or similar sizes.
- Performs a regression analysis for the binned HESs and their average ranks.
- Assigns the estimated HES to all compounds in the HEA by regression and integrates the HESs of all HEAs by summation for every compound.
BEAR’s performance was then evaluated using a large set of ligands for more than a thousand targets. The authors used different cross-validation schemes to evaluate BEAR’s performance robustly and unbiasedly. The findings were remarkable as BEAR performed well in predicting across different scaffolds and assay sets and was not heavily biased across various drug target families. Its performance was pretty stable between 50 and 1000 input ligands, and contrarily, the number of HEAs positively influenced the performance. BEAR was also compared with a few conventional machine learning methods and clearly outperformed them in predicting drug-target interactions for eight ABC transporters. Finally, the BEAR-predicted candidates of P-gp and BCRP dual inhibitors were validated using in vitro assays, and 9 out of 72 dual inhibitors were confirmed.
Implications and Conclusion
BEAR has several key benefits over traditional VS approaches, including not requiring structural information of either the ligand or the target, allowing resuing of bioassay data, predicting (identifying) a diverse set of candidates, and actively using ‘negative data.’ Given BEAR’s incredible performance, which was even experimentally validated, it has great potential in identifying novel and potent dual inhibitors without relying on any structural information. Despite its astonishing performance, BEAR is limited in that it cannot be used for novel targets with few or no known ligands. It also predicts candidate compounds within the boundaries of the compounds included in the bioassay datasets. Regardless of these drawbacks, BEAR has the potential as a crucial tool for drug discovery, particularly for identifying selective inhibitors by counter-screening against unwanted targets.
Article Source: Reference Paper
Learn More:




Diyan Jain is a second-year undergraduate majoring in Biotechnology at Imperial College, London, and currently interning as a scientific content writer at CBIRT. His passion for writing and science has led him to pursue this opportunity to communicate cutting-edge research and discoveries engagingly to a broader public. Diyan is also working on a personal research project to evaluate the potential for genome sequencing studies and GWAS to identify disease likelihood and determine personalized treatments. With his fascination for bioinformatics and science communication, he is committed to delivering high-quality content a CBIRT.





{kind=link}