
A web-based tool called Protein Interaction Explorer (PIE) by researchers from Université de Paris, France, was created to support structure-based drug discovery efforts that center on protein-protein interactions (PPIs). PIE is integrated into the database iPPI-DB. Researchers can detect and characterize important variables such as binding pockets, functional binding sites, hot spots, and protein-embedded pockets for possible repurposing efforts with the help of PIE’s suite of tools for PPI drug discovery.
Introduction
Protein-protein interactions (PPIs) have a major function in drug development and are an essential component of biological pathways and cellular processes. Their potential for small molecule binding and biological function is influenced by their three-dimensional (3D) structure. The term “ligandability” refers to the requirement that a ligandable binding site exists inside the target, underscoring the significance of PPIs in biological pathways and cellular functions.
Protein-protein interactions (PPI) binding sites vary widely in number and size, so assessing their ligandability requires careful inspection. To visualize PPI networks and help with gene-based clustering, route detection, functional module identification, and disease mechanism comprehension, web-based applications such as PIMA, OpenPIP, Cytoscape, Proteinarium, and Cellmap have become popular. Other tools that apply geometric analysis to find protein surface cavities, pocket descriptors, and druggability predictions include SiteMap, DoGSite3, PLIC database, P2Rank, Coach, and Cofactor. Unusual features, including interactive study of protein-ligand binding interfaces and alignment and similarity assessment of protein-protein interfaces, are provided by more recent tools like PiMine and ProteinsPlus. A chance to investigate different strategies for focusing on these interactions with small-molecule inhibitors exists despite difficulties brought on by the absence of easily accessible ligands for PPIs.
Understanding Protein Interaction Explorer (PIE)
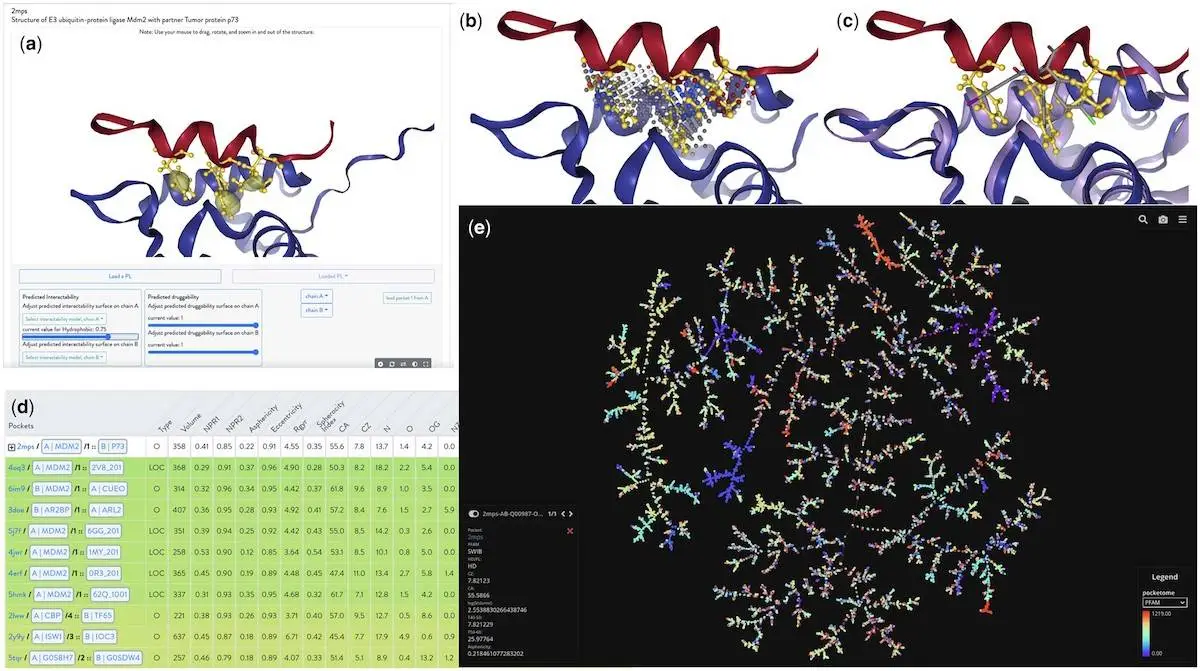
The Protein Interaction Explorer (PIE) is a novel web-based tool that focuses on protein structures, specifically heterodimer complexes. It aids in identifying functional binding sites, predicting hot spots, and visualizing protein-ligand interactions. PIE offers 3D visualization of druggability and intractability predictions and a graphical representation of the “pocketome” derived from the IPPIDB database. It integrates various visualization tools into a unified platform, allowing users to explore PPIs and their modulators. PIE also introduces unique features like the integration of detected pockets through VolSite, functional binding site predictions from InDeep, calculated hot spots using FoldX, and a novel metric for pocket similarity.
Potential Applications of PIE
PIE is essential to structural biology and bioinformatics because it makes it possible for scientists to investigate and examine the complex realm of protein interactions and structures. It helps to progress drug discovery, disease knowledge, and molecular biology by bridging the gap between structural data and biological insights. PIE offers protein structures and interactions, data integration, accessibility, and visualization, allowing researchers to confirm discoveries, work together, and expand on prior knowledge. Because of its intuitive interface, which facilitates information exchange and collaboration, it is possible to compare analyses across various proteins and complexes. By utilizing InDeep predictions, PIE facilitates the evaluation of ligandability by pinpointing druggable areas and binding sites within proteins. Insights for medication design and treatment targeting are also obtained by predicting hot zones.
Detection of Pockets
Protein targets and their related partners were randomly selected for pocket detection and characterization using VolSite. The protein target and its ligand were shown to contain pockets in HD complexes, whereas the monomer was found to contain pockets in PL complexes. Only pockets that fulfilled certain requirements—such as having four or more probes in their negative picture situated one atomic distance apart from a partner—were kept after detection. Based on the ligand’s proximity to its protein partner, orthosteric pockets were classified as either non-competitive or competitive.
The study identified three types of PL pockets: allosteric (PLA), orthosteric non-competitive (PLONC), and orthosteric competitive (PLOC). In PLOC pockets, the epitope of the protein partner and the ligand compete directly within the heterodimer. Ligands found in orthosteric pockets within PLONC pockets have the potential to affect the conformation or function of the protein. PLA pockets may produce allosteric effects when positioned close to orthosteric binding pockets of a heterodimer. These pockets do not directly overlap with the orthosteric site. Using VolSite software, 89 pocket descriptors were calculated. Ten additional descriptors were combined to produce a more complex representation of pocket characteristics. The TMAP tool was used to portray the pocketome as a minimum spanning tree visually.
Exploring Interactions with PIE
Comprehensive predictions and illustrations of the p53-bound MDM2 pocket offer a refined and adaptable examination of hydrophobic channels. A thorough investigation of the binding modalities of various ligands and their chemical moieties may be conducted thanks to the system’s visualization in the NGL javascript window, FoldX hot spot predictions, and VolSite pocket. Additionally, neighboring pockets are displayed using PSI metrics in the visualization; larger values denote a higher degree of pocket resemblance. Other non-MDM2 systems that share similarities with the p53 helix that links the MDM2 pocket with hydrophobic residues include the histone lysine acetyltransferase CREBBP, WD_REPEATS_REGION domain-containing protein, and ADP-ribosylation factor-like protein 2-binding protein. Another method of evaluating the outcomes is to look at the pocketome’s TMAP visualization, which considers the number of common neighbors in the tree.
Conclusion
PIE is an invaluable tool for molecular biologists to investigate protein structures and molecular interactions. It is a vital tool for molecular biology, drug development, and illness understanding because of its intuitive interface, prediction tools, data integration, and customization options. PIE provides an extensive toolkit for analyzing and visualizing complicated interactions, eventually leading to a deeper knowledge of the molecular underpinning of life. As a result, PIE considerably impacts the understanding of molecular interactions and protein functions.
Article Source: Reference Paper | PIE is user-friendly and readily accessible at https://ippidb.pasteur.fr/targetcentric/.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











: a robust platform for exploring protein-protein interactions and ligand binding pockets.){kind=link}