
The availability of genome data is of great importance in this day and age. But, transcriptome data (which is a subset of the entire genome, a set of transcribed genes into RNA) is more readily available than the entire genome data for many species. Yet, it has limitations since it always needs a reference set of genes for gene calling, especially for non-model organisms- where the absence of a reference genome complicates the process and often leads to inaccurate results due to the high redundancy in de novo transcriptome assemblies. To address this issue, a team of researchers from Michigan State University has developed UnigeneFinder, an innovative automated pipeline designed to enhance the accuracy and efficiency of gene calling from transcriptome assemblies without requiring a reference genome.
The Challenge of Gene Calling Without a Reference Genome
Gene calling, or the process of identifying regions of genomic DNA that encode genes, is a fundamental task in genomics. When a reference genome is available, this process is pretty easy. However, for many species, especially non-model organisms (species not commonly used as a standard reference in research), a reference genome is either unavailable or incomplete. In such cases, researchers rely on de novo transcriptome assemblies to infer gene sequences.
De novo transcriptome assemblies, though valuable, come with some challenges. One of them is redundancy, where multiple transcript variants are assembled for the same gene, leading to an inflated number of transcripts. This redundancy can obscure the true gene content and hinder downstream analyses. Existing tools often require manual intervention or multiple steps to reduce redundancy, making the process prone to errors.
Introducing UnigeneFinder
Now, scientists have found a solution to these problems! This new method, allowing multiple gene clustering techniques, significantly reduces the expected redundancy from raw transcriptome assemblies, resulting in a lean and accurate representation of the underlying genome.
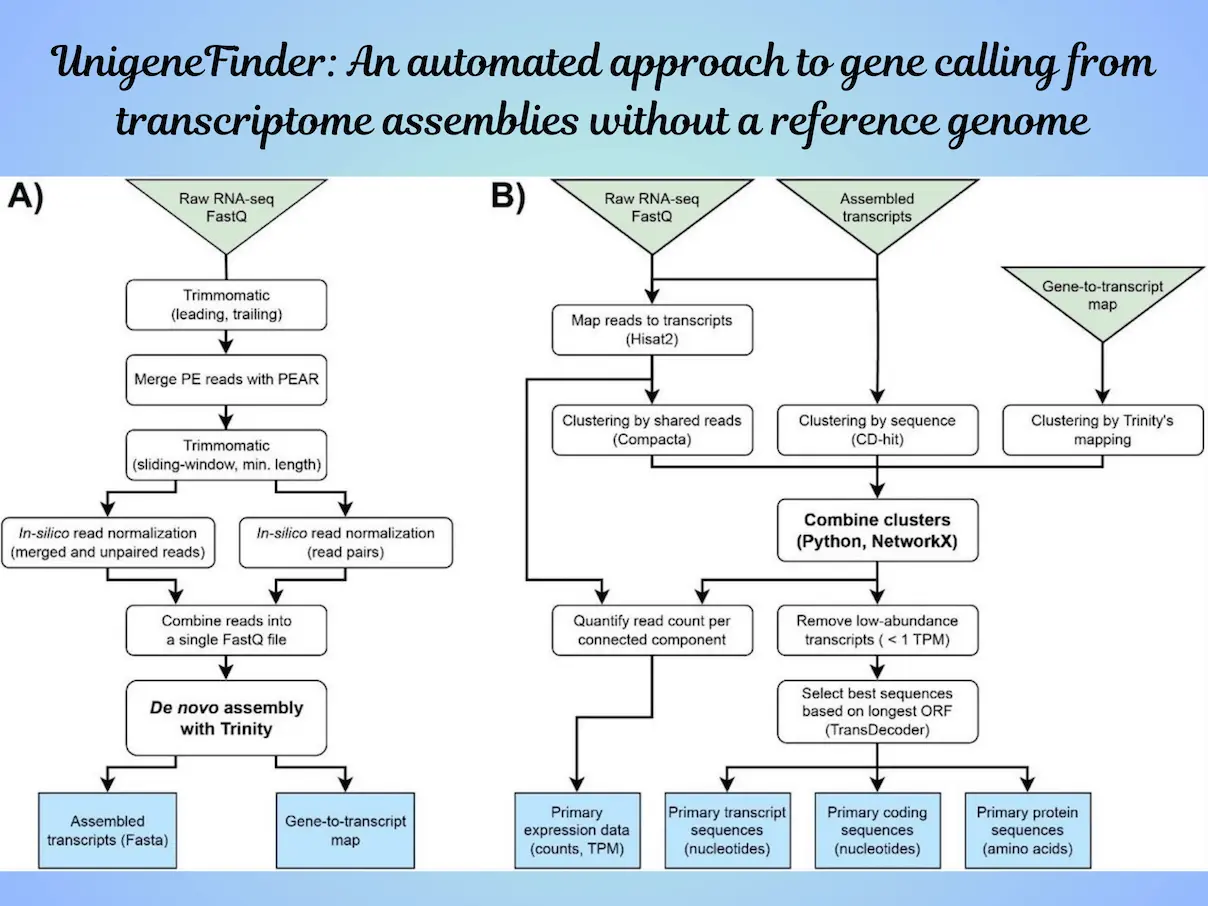
It uses the Python framework. It is user-friendly to advanced computer users, cross-platform, and can run on personal computers, thus making it available to a larger number of researchers. The basic input to UnigeneFinder is a transcriptome assembly in Fasta format, but more accuracy can be achieved by providing FastQ read files or BAM format read mappings. If the mapping of reads is unavailable, the pipeline incorporates a script, unigenefinder_map.py, which does the mapping. This makes it easier for the user.
How UnigeneFinder Works
Thus, in essence, UnigeneFinder is a stepwise process aimed at refining the input transcriptome assembly and reducing redundancy. It starts with the generally generated primary transcriptome assembly based on de novo assemblers, particularly Trinity. This typical primary assembly contains a large number of redundant transcripts, which are then fed into UnigeneFinder for processing and consolidation into unigene sequences.
The performance of UnigeneFinder is based on its implementation of multiple clustering approaches within the same component. This way, the inflated numbers of transcripts will be downsized to get a better representative view of the gene content. The method is not only beneficial in processes gene calling but also enhancing the accuracy of the gene set that is finally achieved, and it will be compared to be equal or, in some approaches, better than the existing applications done recently by these tools.
Performance and Benefits
The performance of UnigeneFinder was thoroughly benchmarked using different plant species when considering varying levels of genome complexity, varying from simple to complex. As has been shown from the results, UnigeneFinder, in terms of precision, is approaching the existing tools for gene finding at par or even surpassing most of them when considering the F1-scores, which equally measure differences in the trade-off between precision and recall for genes’ identification. One of the remarkable virtues of UnigeneFinder is that it can easily and fully automate the generation of primary sequences, similar to high-quality reference genomes, for transcripts, coding regions, and proteins!
The tool can equally be used in such a way that it can help even a user who has little experience in computation to generate trustworthy gene sets using the transcriptome data.
In fact, the more improved and affordable sequencing technologies there are, the more volumes of transcriptome data will be increasingly available for non-model organisms. Tools such as UnigeneFinder will filter down negatively and frantically huge data and, in essence, they will enable researchers to keep up with the rapid growth in available information. This is a wonderful invention for the future of genomics!
Conclusion
With accuracy, automation, and ease of use, UnigeneFinder stands out for gene calling without using a reference genome. In this way, UnigeneFinder can go a long way to begin to overcome these twin problems and open up the study of non-model organisms, increasing the understanding of the genetic diversity of life.
Article Source: Reference Paper | UnigeneFinder is available on GitHub
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Neermita Bhattacharya is a consulting Scientific Content Writing Intern at CBIRT. She is pursuing B.Tech in computer science from IIT Jodhpur. She has a niche interest in the amalgamation of biological concepts and computer science and wishes to pursue higher studies in related fields. She has quite a bunch of hobbies- swimming, dancing ballet, playing the violin, guitar, ukulele, singing, drawing and painting, reading novels, playing indie videogames and writing short stories. She is excited to delve deeper into the fields of bioinformatics, genetics and computational biology and possibly help the world through research!











{kind=link}