
A prevailing challenge in genomics still revolves around the comprehension of structural variants, one of which is copy number variation (CNV). To undertake the task of conquering this challenge, researchers from the National Institutes of Health, along with others, have developed a groundbreaking software known as CNV Finder. This deep-learning-based software assists in CNV identification and aids in redefining the entire process. This state-of-the-art pipeline delivers a quick, straightforward CNV solution that can be extended on several related factors, like the CNVs about the region 17q21.31, which is also known for containing the MAPT gene and other complicated haplotypes associated with it.
The Complexity of CNVs in the 17q21.31 Region
This part of the chromosome is genomically active but very complex owing to the structural variation caused by certain haplotypes, such as the H1 and inverted H2 branched regions. As a result, this area has been associated with various neurological diseases, including Alzheimer’s disease (AD), progressive supranuclear palsy (PSP), and other tauopathies. Considering this, the deletions between 461 and 463 kilobases on chromosome 17 in the KANSL1 region might have been accepted as the most common CNV.
Traditional CNV detection methods often falter in these regions due to sparse or noisy data from genotyping arrays. However, the lack of inborn assistance by the software providers can be balanced or rather compensated for through the augmentation of computer models and the validation of experts, enabling smoother analysis and, ultimately, identification of CNV.
How CNV-Finder Works
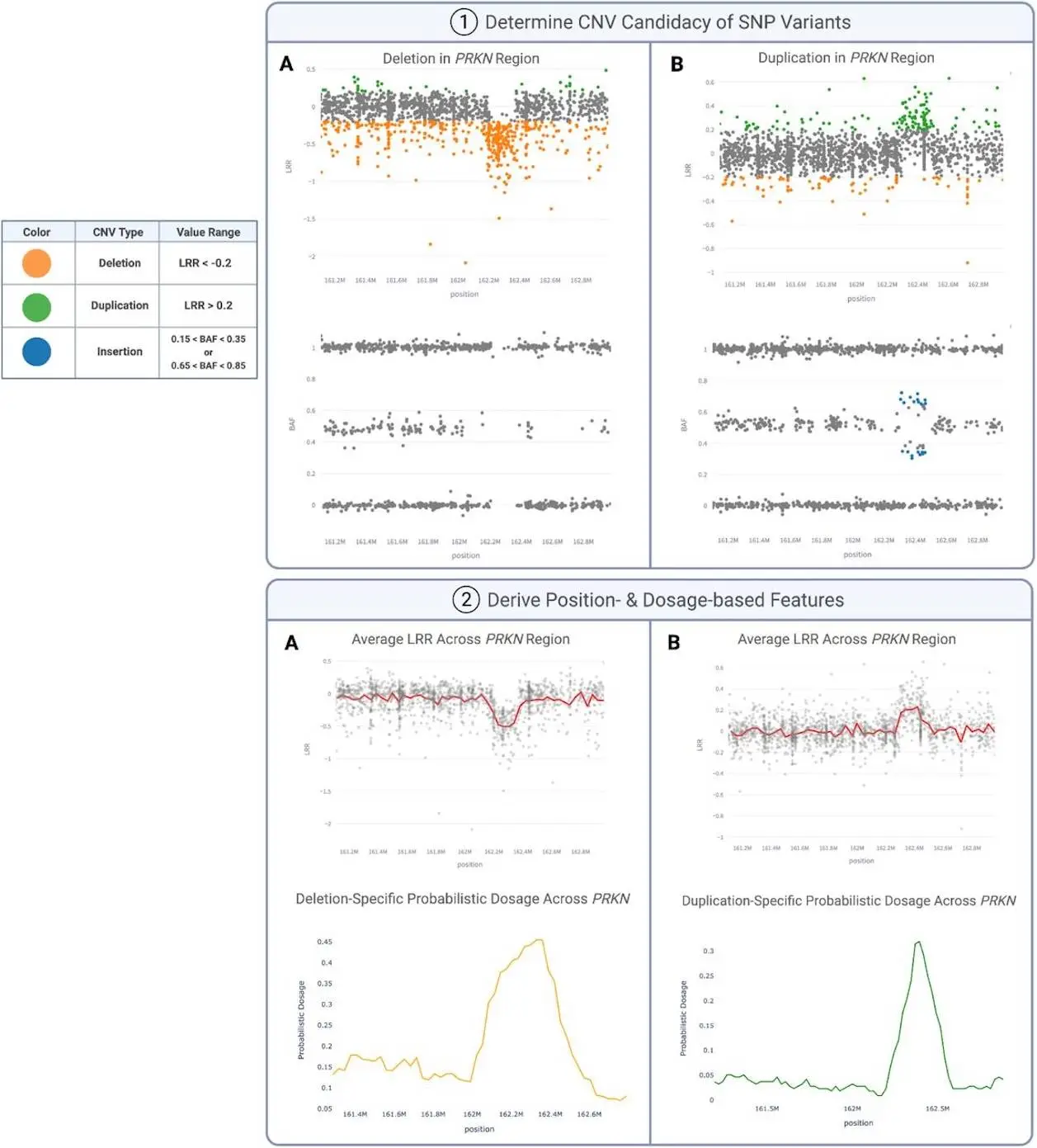
The deep learning model-enabled CNV-Finder uses a semi-automated pipeline. A sequence-to-vector deep learning LSTM (Long Short-Term Memory) framework is employed to quickly locate the positions of the CNVs in more than preset genomic regions. For the purposes of the model pipeline training, the dataset contained samples marked as Yes, Maybe, and No depending on the presence of CNV, enabling the pipeline to evaluate the veracity of the CNVs, cases of uncertain veracity, and CNV artifacts.
Regions like the 17q21.31, where duplications can colocalize with signal artifacts, require some level of human input. Researchers manually reviewed array-based plots—such as log R ratio (LRR) and B-allele frequency (BAF) vs. chromosome position—to flag cases for additional validation. Such meticulous curation ensured that borderline cases were neither too hastily classified to be true CNVs nor excluded from the training datasets.
Insights into H1 and H2 Haplotypes
A major finding of the study was the strong association between CNVs and the H2 haplotype, characterized by tagging SNP rs8070723. Out of the 1151 samples included in this study, the researchers reached an astonishing similarity score of 0.94 using the Jaccard test for H2-carrier and CNV presence. Logistic analysis also reinforced the idea that 97% accuracy with 0.95 AUC could be attained just by determining whether or not the carrier status of the H2 haplotype had duplication.
Interestingly, it appears that H2 non-carriers (H1H1 haplotype) had fewer duplication events in the MAPT region. While in cohorts such as MDGAP-DEMENTIA and REGARDS, the model seemed to suggest duplications of 39 types in H2 carriers only, and a composition of 64% H1H1 in those cohorts was also noted. This qualifies as an important observation that most probably explains the complexity of the relationship between the haplotype structure and CNV detection.
Challenges and Solutions
Despite its strengths, CNV-Finder isn’t without limitations. Human validation, while essential for enhancing model accuracy, can introduce inconsistencies and bias. Furthermore, it seems to pose a challenge to have an appropriately balanced training dataset comprising the requisite volume of both positive and negative CNV samples, especially with the growing improvement in the diagnosis.
In addition, the study stated that the technological limits of array-based methods were commendable relative to high-resolution techniques like the long-read sequencing technology. For instance, in some cases, such as Sniffles2, the program got stuck in the region of the MAPT and could not locate the duplication breakpoints because the chromosome lost CFU after the CNV event. Even using genes by themselves for mapping crossovers, some have acknowledged that crossover starts occur sooner than loci specify.
Conclusion
The study’s findings stress the need for blending human skills and advanced computational models. As CNV-Finder continues to advance toward broad applications across the entire genome and higher resolution sequences, its value in clinical and research genomics is expected to increase rapidly. With the conquering of regions such as the 17q21.31, CNV-Finder not only assists in comprehension of the genetics of populations but also paves the way into personalized medicine.
In a world where each new finding is one step further into understanding the genetic causes of most diseases, CNV-Finder ranks as the most novel finding in the sense that it enables temporally the extraction of meaning from the raw data.
Article Source: Reference Paper | The CNV-Finder pipeline is a scalable, publicly available resource for the scientific community, available on GitHub.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}