
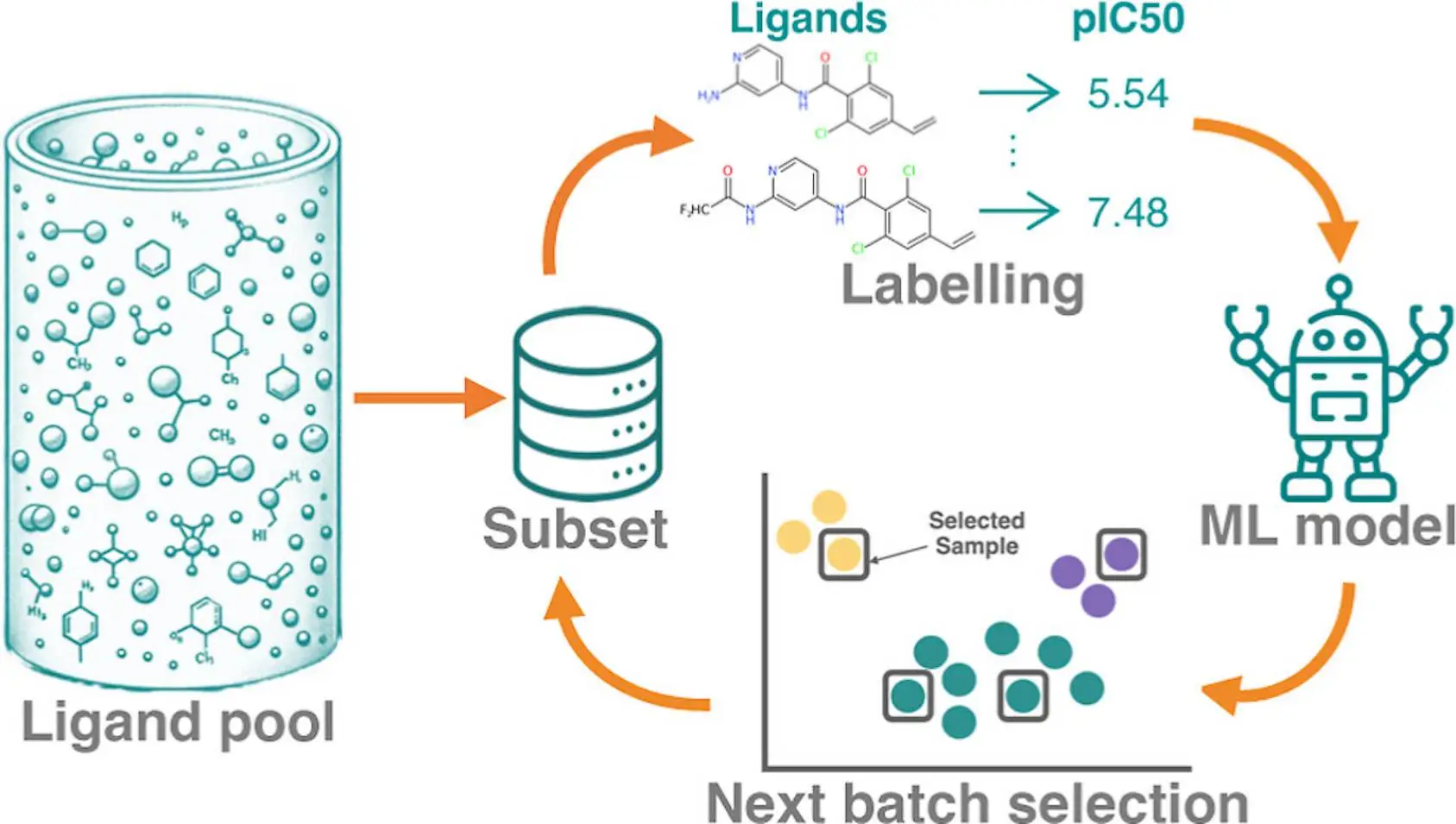
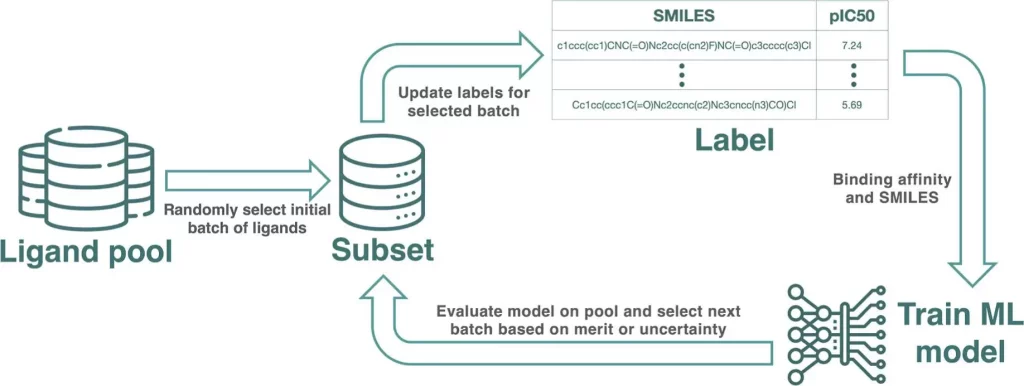
In computational drug discovery, active learning (AL) is a potent approach that makes it possible to identify the best binders from large chemical libraries. To get the best results, designing a strong AL methodology necessitates an understanding of AL parameters and data set properties. In order to systematically assess the efficacy of machine learning models [Gaussian process (GP) model and Chemprop model], sample selection protocols, and batch size, researchers use four affinity data sets for distinct targets (TYK2, USP7, D2R, and Mpro). These metrics describe the overall predictive power of the model (R2, Spearman rank, root-mean-square error), as well as the accurate identification of top 2%/5% binders (Recall, F1 score). On large data sets, the recall of top binders for both the Chemprop and GP models is comparable; however, given sparse training data, the GP model performs better than the Chemprop model. Although smaller batch sizes are favored for subsequent cycles, larger initial batch sizes boost recall and correlation measures. While clusters containing high-scoring chemicals can be found using artificial Gaussian noise, too much noise reduces the ability to foresee and take advantage of opportunities.
What is Active learning?
Active learning is a semisupervised machine learning technique that chooses fresh samples to identify unlabeled data iteratively. This approach is especially helpful in computational drug discovery as it can find potent inhibitors in small-molecule libraries at a lesser cost than a systematic potency screen. The procedure entails striking a balance between the optimization of known leads through slight substitutions and innovation from exploring a new chemical space. In order to pick samples representative of the underlying chemical space, AL campaigns frequently combine exploration and exploitation tactics. The exploitation strategy retrieves a large number of potent compounds based on projected binding affinity.
Alchemical Free Energy (AFE) methods have historically been employed in lead optimization to choose compounds for synthesis in the latter stages of the process. These techniques, however, are generally linked to a limited chemical space and have poor throughput. Computational potency prediction tools have developed from classical docking, AFE methodologies, and ML approaches during the past forty years. Due to the increase in automation and throughput of computational tools for drug discovery, the use of AL applications and computational potency estimation, such as virtual screening or relative binding free energy (RBFE) calculations using molecular dynamics simulations, has only become more prevalent in the last eight years.
Compared to low-throughput use cases, the compound pool’s sheer vastness and a high degree of diversity place more strain on the AL pipeline and call for careful consideration when choosing molecular characteristics, machine-learning models, and acquisition techniques. Furthermore, systematic and stochastic mistakes are introduced when docking scores or RBFEs are used instead of experimental binding affinities. These flaws are frequently poorly defined beforehand.
Setbacks Faced by Active Learning
In the pharmaceutical sector, regular use of AL necessitates a reliable technique that can be applied to various data sets. Prior AL research has made use of RBFE data; however, there are obstacles due to the lack of big public RBFE data sets and the expense of creating them. The selection of protocols is done without taking cost into account, which leads to big first batches or experimentation stages. Protocols that demand different quantities of RBFE data also vary. The variances in data sets, sizes, generation processes, and implemented AL protocols pose challenges to the comparison of literature protocols and the identification of optimal methods for the regular implementation of AL in the pharmaceutical sector.
Goal of the Study
This study aims to evaluate AL protocols in a rigorous manner using four publicly available datasets for benchmarking. The AL design is the primary focus, not the labeling technique employed. An ML property prediction model, AFE techniques, experimental measurements, and docking are a few different labeling options. The study makes use of four distinct labeled data sets that differ in terms of size, degree of variety, protein targets, and potency measurement. The main goal is determining how the number and diversity of data sets impact AL’s effectiveness.
To provide a comprehensive view, a variety of metrics are employed, such as pre-trained model architectures for AL, recall and F1 scores for the top 2% and 5% of binders, and traditional regression metrics. In order to compare AL procedures at a consistent cost, the total number of obtained samples is maintained constant throughout all tests. Any labeling technique, including docking score, experimental value, and RBFE computation, can be applied to prelabeled data. However, combining various approaches necessitates careful consideration of accuracy or reliability, which is outside the purview of this work.
Exploring Benchmarking Active Learning Protocols
The purpose of this study was to compare the performance of two models on every data set in a sizable training set. The findings demonstrated that the training sets only made up a small portion of the pool; following AL experiments, less than 20% of the whole data was obtained, with the exception of Mpro, where more than half of the data was obtained. Given a substantial quantity of training data, the analysis yielded an upper bound for the model’s regression performance as well as an estimate for recall. With an R2 greater than 0.3, a Spearman ρ greater than 0.5, and top 5% recalls of 0.2 or above, all models showed promise as predictors.
However, there was a noticeable pattern in the model’s performance with respect to the size of the data set: R2 and top 5% Recall monotonically decreased as the data set shrank. Recall diminishing monotonically as the size of the data collection decreases. The diverse character of the Mpro and D2R data sets posed difficulties because of their lower Spearman ρ and lack of identifiable grouping in UMAP projections. It appears that chirality representation did not considerably improve the models’ performance since the GP model trained on achiral fingerprints performed similarly to the model employing chirality descriptors.
Conclusion
The study assessed how various Artificial Neural Network (AL) protocol settings affected metrics like the identification of top binders, the ML model’s prediction quality, and the qualitative examination of chemical space clusters. The outcomes demonstrated that AL, with a clear influence on the size of the data set, can find top binders with a reduced fraction of data used for training. The most important aspect influencing the margin of profit that AL can achieve is the diversity of compounds within a data set. The study also discovered that specific design techniques can aid in creating the most beneficial AL protocols, which has consequences for how ligand pools are created for AL. Additionally, the study discovered that while performance gains are incremental when batch size is lowered below 30 samples, training in small batches yields the highest recall. The exploiting power of AL was negatively impacted by the addition of noise to potency data, whereas the GP model identified large-scale active zones in the chemical space.
Article Source: Reference Paper | All the data used in this study can be found on GitHub
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}