
A notable progress in protein prediction and generative tasks, such as 3D structure prediction, protein design, and conformational dynamics, has been made in recent years with the rapid development of protein foundation models. Nevertheless, the lack of a consistent evaluation methodology means that the potential and constraints of these models are still little understood. Researchers have presented a comprehensive assessment system called ProteinBench to close this gap and improve the transparency of protein foundation models. The goal of the publicly available toolkit ProteinBench is to create a uniform framework for evaluating protein foundation models, promoting cooperation among researchers, and advancing the field’s creation and use of these models. The thorough analysis of protein foundation models yields a number of important conclusions that clarify the current state of play and the limitations of these models. Researchers make the assessment dataset, code, and public leaderboard available to the public for additional analysis and the creation of a general modular toolset in order to encourage openness and support future studies.
Introduction
Proteins are essential molecules that are essential to many biological activities, including signal transduction, enzyme catalysis, immunological response, and structural support. Their amino acid sequences, which are frequently mediated by folding into particular three-dimensional structures, dictate their functions. For the fields of pharmaceuticals, agriculture, specialty chemicals, and biofuels to advance scientifically and technically, it is essential to comprehend the intricate interactions among protein sequence, structure, and function.
Diving Deep Into Foundation Models
The creation of protein foundation models, which attempt to capture the complex operations of proteins in order to comprehend basic biological processes, has increased dramatically in the last several years. With the use of cutting-edge deep-learning and generative AI techniques, these models have shown impressive capabilities. This represents a major departure from traditional, task-specific approaches and a move towards more generalizable frameworks that can recognize intricate patterns and relationships within sizable protein datasets. While some, like the ESM series and DPLM, have demonstrated impressive representation capability in protein language modeling, which benefits a variety of downstream tasks, others, like AlphaFold3, which is based on diffusion models, have achieved unprecedented accuracy in full atom structure prediction for all biomolecules. Moreover, these fundamental models do not confine themselves to a particular modality.
To provide a thorough knowledge of protein behavior, multi-modal models that take sequence, structure, and function into account are beginning to emerge. Understanding the conformational dynamics of proteins is crucial to comprehending the link between sequence, structure, and function. To simulate the conformational distribution of proteins, generative artificial intelligence (AI) has been established, and recent work has expanded protein structure prediction to numerous conformation prediction tasks.
The necessity for a consistent framework to comprehensively assess the performance of protein foundation models across a wide range of tasks, datasets, and metrics has arisen, despite the models’ rapid advancement. Modeling methodologies that are not unified and evaluation criteria that are specific to individual tasks or models characterize the state of protein foundation models today. Since assessment techniques vary widely, it can be difficult to completely comprehend the relative advantages and disadvantages of various models and to make meaningful comparisons between them.
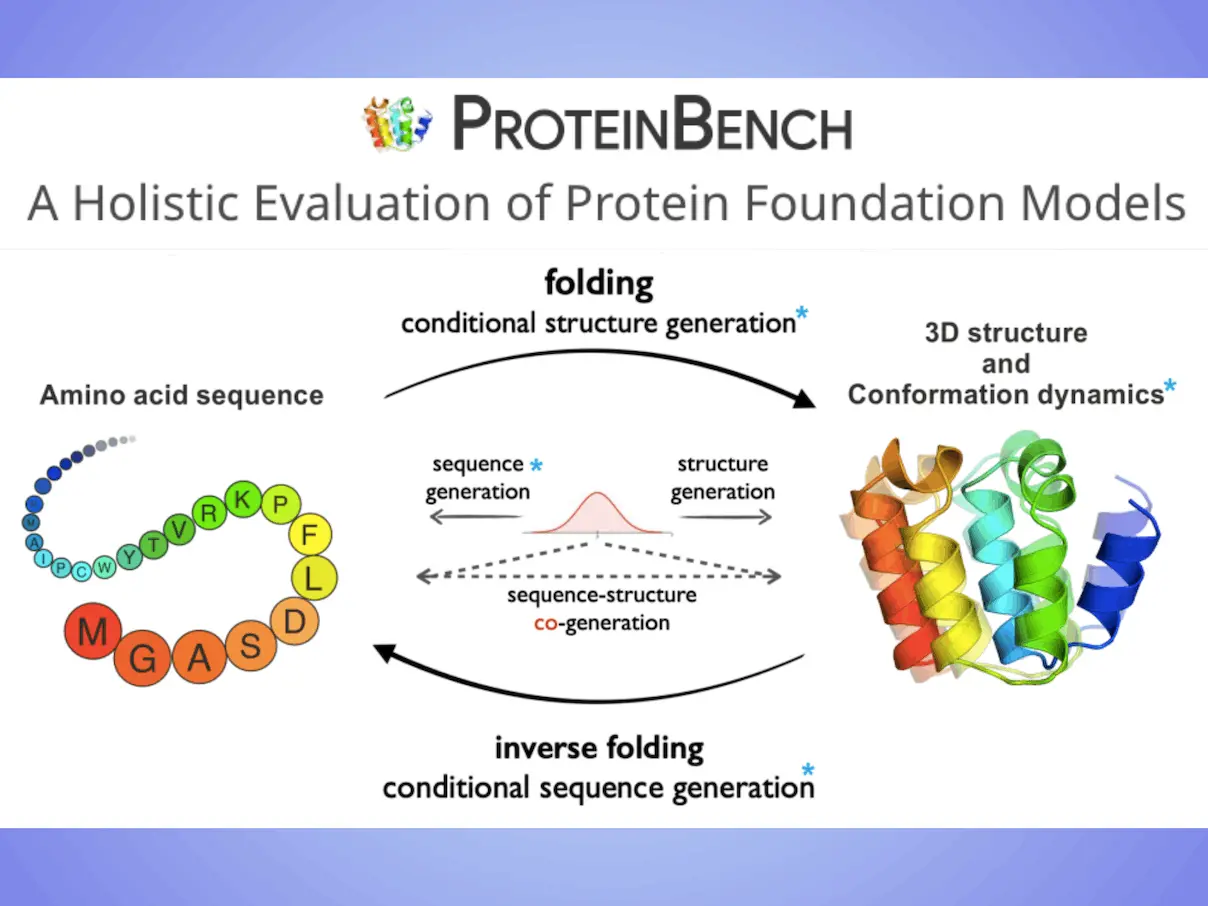
About ProteinBench
Scientists introduce ProteinBench in this work. The researcher’s objective is to offer a thorough examination of model architecture and performance on protein foundation models using methodical assessments of datasets that include several biological areas, with a specific focus on protein design and conformational dynamics. They analyzed the effects of different model elements and data features on distinct facets of protein modeling with this method. For the purpose of directing future research paths, selecting models for real-world applications, and advancing the field overall, it is imperative to compare these models’ capabilities on standardized benchmarks. The strategy is comprised of three main parts: (i) Based on the relationships between various protein modalities, a taxonomy classification of tasks that broadly encompasses the main challenges in the protein domain; (ii) A multi-metric evaluation approach that evaluates performance across four key dimensions: quality, novelty, diversity, and robustness; and (iii) In-depth analyses from various user objectives, offering a comprehensive view of model performance.
Setbacks and Future Work
Researchers note several shortcomings and areas where the present benchmark should be improved:
- There might not be a complete list of basis models chosen. To provide a more thorough comparison, future editions should include more foundation models.
- Currently, direct comparisons of alternative model designs are hampered by inconsistencies in training data among models. This could be fixed in the future by standardizing datasets, which would enable more precise architectural performance comparisons.
- A larger variety of jobs might be included to increase the benchmark’s usefulness and scope. Researchers are dedicated to improving and growing ProteinBench throughout time. The goal is for it to develop into a dynamic, expanding benchmark that spurs advancement in the field of protein modeling and design.
Conclusion
The first thorough analysis of the performance of different protein foundation models on eight different tasks is presented by researchers, with an emphasis on protein design and conformation dynamics. To evaluate protein foundation models objectively from a variety of perspectives, scientists have created a single, multi-metric assessment methodology. Researchers share insights and considerations for the creation and efficient application of protein foundation models, providing direction for future study, based on the performance outcomes. A thorough analysis of the current protein foundation model benchmarks is given, highlighting the scope and application constraints of these benchmarks. These benchmarks mostly address task-specific assessments, ignoring important facets such as sequence design and structure-sequence co-design. One notable feature of ProteinBench is its extensive coverage of a wide range of tasks, such as single-state folding and multiple-state prediction, inverse folding, backbone design, sequence design, structure-sequence co-design, antibody design, and so on.
Article Source: Reference Paper | Project Page: GitHub
Important Note: arRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}