
In recent years, deep learning algorithms have shown promise in identifying protein function and extracting complex sequence-structure-function correlations.

Image Source: Learning Functional Properties of Proteins with language models
Scientists from Middle East Technical University and Karadeniz Technical University present a benchmarking study that compares the results and benefits of various deep learning algorithms in various protein prediction tasks.
The data-centric approaches have been utilized for the development of predictive methodologies for explaining uncharacterized properties of proteins; in any case, studies show that these strategies ought to be additionally improved to tackle critical problems in biomedicine and biotechnology, which can be accomplished by better addressing the current data.
Novel approaches to data representation, for the most part, take motivation from language models that have yielded notable improvements in natural language processing. Of late, these methodologies have been applied to protein science and have shown profoundly encouraging results in terms of extraction of complex sequence-structure-function relationships.
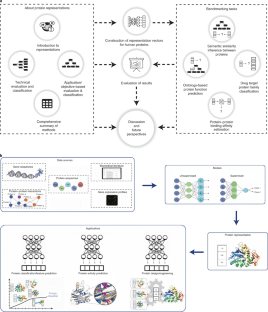
In this study, the scientists conducted a point by point investigation over protein representation learning by first categorizing/elucidating each methodology, along these lines benchmarking their performances on the prediction of:
- Semantic similarities between proteins,
- Ontology-based protein function,
- Drug-target protein families and
- Protein-protein binding affinity changes followed after mutations
The scientists evaluated and discussed the benefits and disadvantages of every strategy over the benchmark outcomes, source datasets, and algorithms utilized in close comparison with conventional model-driven approaches. Finally, the researchers discussed current difficulties and recommended future bearings.
They believed that the conclusions of this study would assist researchers with applying machine/deep learning-based representation techniques to protein data for different predictive tasks and rouse the improvement of novel approaches.
Protein Science
Protein science is known to be a broad discipline that makes analyses of both individualistic and the whole proteomes of living organisms. It utilizes laboratory experiments or proteomics and computational techniques like machine learning, molecular modeling, data science, etc., to create high-accuracy re-usable methods, which are highly revered and utilized in biotechnology and biomedicine.
Protein Informatics
Protein informatics is referred to as an extension of protein science that mainly deals with the modeling of quantitative aspects of proteins in a computationally data-centric manner.
The functional characterization of proteins is critical for the development of new and efficiently effective biomedical strategies and the products of biotechnology. The number of protein entries since May 2021 was known to be around 215 million in the UniProt protein sequence and annotation knowledge base, and of those, only 0.56 million are known to be manually reviewed with annotation expert curators, which indicated the large gap between the current sequencing and annotation capabilities.
The gap is solely due to the cost required and the time-consuming nature of acquiring results from wet-laboratory experiments and the manual curation later on.
For the augmentation of experimental and curation-based annotation methodologies, in-silico approaches have been utilized, in this context, numerous research groups have since long been working on the development of new computational methods for the prediction of enzymatic activities of proteins, biophysical properties, protein-ligand interactions, 3D structures, and finally, their functions.
Protein Function Prediction (PFP)
Protein function prediction or PFP is referred to as the functional definition assignment to proteins, which can be semi-automatic or automatic.
The chief terminology for biomolecule functions can be codified in the Gene Ontology System or the GO system, which is a hierarchical network of concepts or controlled vocabulary which annotates molecular functions of genes and/or proteins, along with their subcellular localizations, including the biological processes in which they are involved.
The most extensive and comprehensive benchmarking project of PFP is known to be the Critical Assessment of Functional Annotation challenge or the CAFA challenge.
Protein Representation Approaches
The protein representation methodologies can be classified into two main categories, which are:
- Classical representations or a model-driven approach: Generated by the utilization of predefined rules about properties like evolutionary relationships between genes/proteins or the physicochemical properties of amino acids.
- Data-driven representations: Constructed by utilization of statistical and machine learning algorithms, which are trained for predefined tasks like a prediction of the following amino acid on a sequence.
After that, the output of the trained model, known as the representation feature vector, can be utilized for other different protein informatics-related tasks, such as function prediction.
In the given sense, the representation learning models influence knowledge transfer from one task to the other.
The generic form of this process is called transfer learning which is analyzed to be a highly efficient and effective data analysis approach in terms of time and cost. Thus, protein representation models reduce the need for data labeling.
The scientists investigated 23 representation learning methods for the benchmark analysis according to the potential of these methods to capture the functional properties of proteins. Aiming to assess the potential of these methods, the benchmarks are constructed and applied based on:
Semantic Similarity Reference
The semantic similarity reference analysis has the objective to quantify the amount of information the representation models capture about the biomolecular functional similarity. The researchers utilized GO annotations representing the molecular functions, large-scale biological roles, and subcellular localization of proteins.
They first calculated the pairwise quantitative similarities between the representation vectors of proteins in their dataset and then compared them with functional similarities between those proteins, which were measured based on actual GO annotations of those proteins by utilizing standard semantic similarity measures.
To compare the success of different protein representation methodologies, the researchers calculated the Spearman rank-order correlation values between representation vector similarities and the actual GO-based semantic similarities of the same protein pairs by utilizing three different test datasets.
In the takeaway, they concluded that the higher the correlation values, the better the success of the representation.
Ontology-Based Protein Function Prediction
The scientists aimed an objective to assess the success of the representation models in the classification-based automated protein function prediction.
In this second benchmark of their study, some learned representation models performed considerably better than some classical methods when analyzed statistically.
The overall performance in some types of GO prediction tasks was lower than that observed in the MF prediction tasks. This was reasonable since most of the learning-based methods make use of protein sequence data as input and the sequence is not a direct indicator for localization or the biological role of protein in large-scale processes.
Drug Target Protein Family Classification
In this benchmark study, the scientists measured the performance of protein representations in the drug discovery framework to predict drug-target proteins’ main families.
As those families are made up of proteins with distinct structural characteristics, this analysis was also expected to reflect the capability of the models in learning the structural properties.
Moreover, by the utilization of a data source other than functional annotations, the scientists sought to diversify their benchmark and evaluate the representation from a different perspective.
The objective behind the benchmarking methodologies over the datasets was to analyze how much the learning was based on simple sequence similarity instead of the learning of complex and hidden patterns corresponding to the prediction tasks at hand.
The results of this study concluded that representation learning methods might have the ability to capture patterns beyond simple sequence similarities.
Protein-Protein Binding Affinity Estimation
In this study, the scientists had the objective to assess the performance of representation methodologies in the prediction of experimentally identified protein-protein binding affinities.
Notably, the change in binding affinities observed due to the occurrence of mutations in one of the interaction partners was predicted in the study.
The scientists utilized the SKEMPI dataset, which contained the PPI binding affinity scores or Kd values between the co-crystallized complexes of both the wild-type proteins and variants.
This study evaluated the representation methodologies in terms of their capability to extract residue and/or region-level structural features, which have critical importance for the physical interactions between protein pairs to occur and how the single, double, or triple amino acid changes affected the binding affinities.
The Endpoint
The quantity of artificial intelligence-based protein informatics studies has been growing in the recent era of computational biotechnology advancements to understand further complex relationships between sequence, structure, and functions.
In this study, the researchers assessed the protein representation learning methodologies to capture the functional properties of proteins to be utilized for the critical challenges in protein science, biotechnology, and biomedicine domains.
Article Source: Unsal, S., Atas, H., Albayrak, M. et al. Learning functional properties of proteins with language models. Nat Mach Intell 4, 227–245 (2022). https://doi.org/10.1038/s42256-022-00457-9
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Tanveen Kaur is a consulting intern at CBIRT, currently, she's pursuing post-graduation in Biotechnology from Shoolini University, Himachal Pradesh. Her interests primarily lay in researching the new advancements in the world of biotechnology and bioinformatics, having a dream of being one of the best researchers.











{kind=link}