
In the ever-changing environment of computational biology, predicting the structure of protein-ligand complexes remains a vital task with far-reaching ramifications for drug development and molecular biology. DynamicBind, a pioneering model described in a Nature Communications paper, is revolutionizing the field of protein-ligand interaction investigations. This novel technique, pioneered by Wei Lu, Jixian Zhang, Shuangjia Zheng, and their colleagues at Galixir Technologies, has enormous potential for drug discovery and structural biology.
The Challenge of Predicting Protein-Ligand Complexes
- Complex Dynamics:
Protein is a highly flexible macromolecule that literally folds and unfolds in the presence of ligands and responds to their binding by changing conformation, which makes it difficult to predict protein-ligand interactions. Cohesive models tend to be static and ignore the fact that proteins are amphipathic, which is a factor that needs to be considered when analyzing protein function.
- Computational Demands:
Molecular dynamics can provide information concerning protein structures, but it is time-consuming due to small high-energy transitions that correspond to biologically significant conformations. This computational complexity becomes a barrier to precise prediction of protein-ligand complex structures, for our gap needs to be filled by innovative approaches.
- The Role of Deep Learning:
Amidst the difficulties in protein-ligand derivative prediction, DynamicBind, a deep learning algorithm, serves as a viable and efficient approach. DynamicBind has the potential to offer new approaches in protein-protein interactions with the help of real-time protein conformations and advanced computational tools that would help in providing a breakthrough into almost every field related to drug discovery and structural biology.
What is DynamicBind?
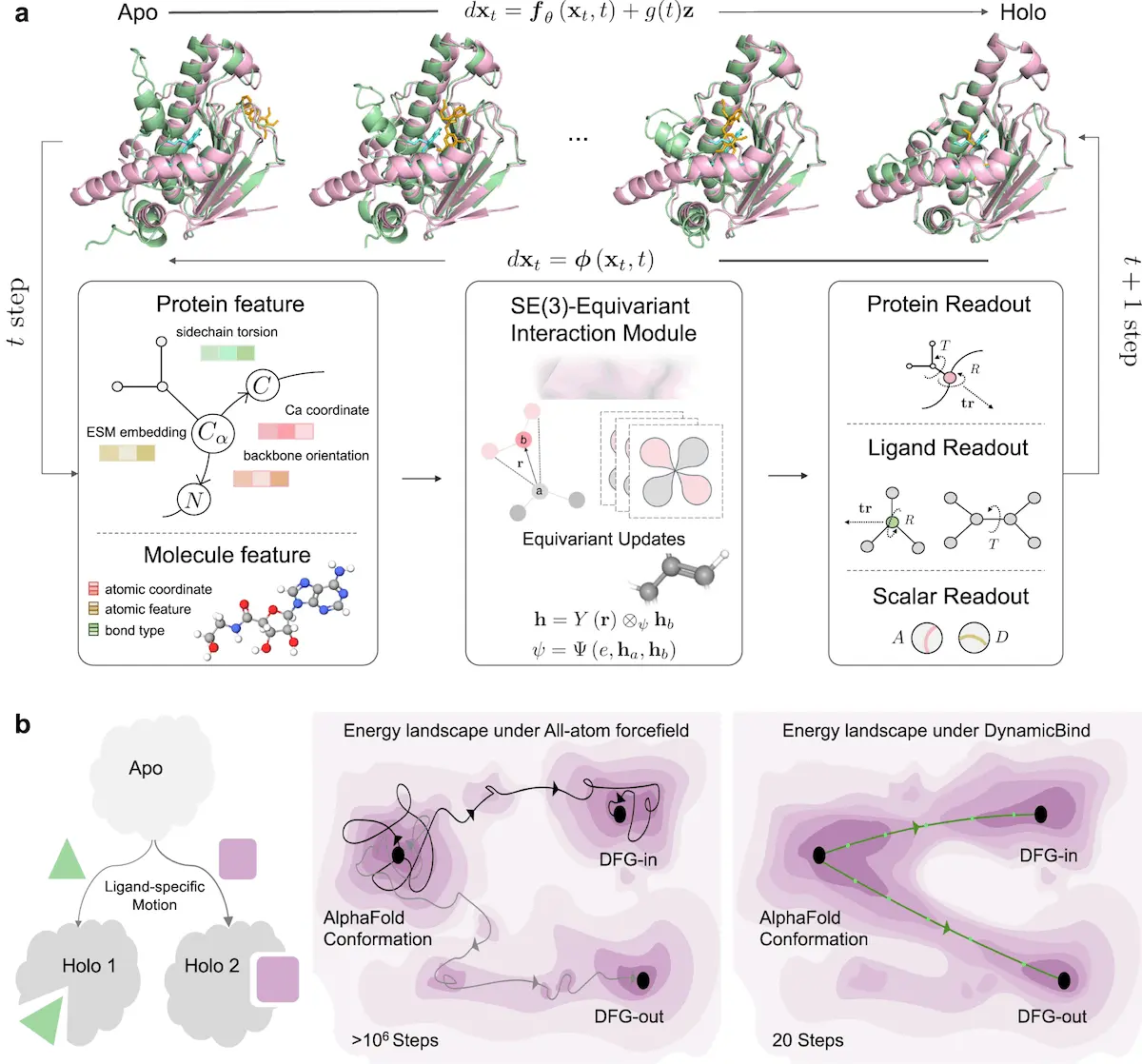
DynamicBind is an extraordinary deep-learning model that introduced a change in protein-ligand interactions by using dynamic docking. It is more effective than other protein binding methods that consider proteins as rigid structures since the approach introduced here can model the dynamic aspect of proteins, making it possible to obtain highly accurate predictions of protein-ligand binding modes. Compared to other methods, this model provides accurate predictions for specific ligand-protein orientations that demonstrate critical insights into the effects of ligands as well as structural and functional relationships in drug design and structural biology.
How DynamicBind Works
DynamicBind works based on deep learning approaches combined with dynamic docking to calculate the interactions between protein and ligand in the shortest possible time and with high efficiency.
i. Dynamic Docking Approach:
By recognizing the movement of proteins, DynamicBind benefits from the flexibility of proteins and the changes in their conformations caused by the ligands. This naturally gives the model a better view of the protein-ligand interactions and structural configurations, which are more realistic.
ii. Training Set Augmentation:
DynamicBind strengthens the base of training data as a by-product of coming up with the extra high-certainty predictions and makes the model stronger and more accurate. Thus, this augmentation strategy makes the model well-prepared to capture or filter details relevant to the intended analysis while avoiding chasing shadows or what is apparent but changes direction when rotated or viewed under a different angle.
iii. Ensemble Docking and Morph-Like Transformations:
Through multi-hit pose generation and morph-like transformations, DynamicBind can effectively sample protein conformational changes and provide an extensive survey over quite diverse timescales necessary to analyze the structures of proteins. This approach not only improves the generalization capacity of the created model but also helps minimize the computational burden typically associated with other approaches.
iv. Deep Learning Model:
DynamicBind can then use deep learning to process and analyze such new and complex input data and train the model to identify details of protein-ligand interactions. Both theoretical and experimental predictions are bridged utilizing complex algorithms through the computational higher-order adaptation of DynamicBind for efficient processes in drug discovery and structural biology applications.
v. Potential for Drug Discovery:
DynamicBind is also useful for mapping unknown binding pockets and exploring ligand-induced protein conformations, which makes it a valuable tool in augmenting drug discovery efforts. In this sense, DynamicBind gives approaches for the strategic and specific medicine associations with the searchlights, which evolved from key structural elements and binding affinities, approaching the way for unique remedies.
Methodology Used in DynamicBind
Use of an E(3)-equivariant and diffusion-based graph neural network where elements are hierarchically diffused using a coarse-grained structure in order to predict protein-ligand interactions with high accuracy and less time.
- E(3)-Equivariant Model:
With trans-rotation and parity operations, the input is mapped to a 3D space, able to facilitate protein dynamics and ligand interactions at the same time, all with the help of the labeled, automated model.
- Diffusion-Based Graph Neural Network:
The author’s combination of the diffusion-based formulation with the graph neural networks, as applied in DynamicBind, can indeed facilitate the identification of the relevant interactions between proteins and ligands and, therefore, predict various aspects of the underlying structural configurations and binding affinities.
- Coarse-Grained Representation:
DynamicBind takes advantage of a large-grained framework of the protein-ligand complexes, allowing the model to encode and learn only the most essential features for prediction while discarding non-critically important nuances that may vary depending on the orientation of the complex.
- Training and Validation:
Multiple curated datasets have been used to train and validate the model, making it possible to demonstrate high performance and transferability to other potential target protein and ligand families.
- Innovative Approach:
Overall, the use of DynamicBind as both an enhanced computational tool and a deep learning-based methodology for predicting protein-ligand binding makes it a valuable addition to the field of research in structure-function relationships, making the drug discovery process more efficient.
Performance and Future Prospects
DynamicBind achieves an accuracy score of 0.764, a success rate that has already showcased the viability of the service in predicting protein-ligand interactions. This is a highly active area of research with enormous opportunities to enhance and develop the model further as several drugs and structural biology projects are in the pipeline, and new therapeutic paradigms are opening up.
Conclusion
In conclusion, the story of DynamicBind is the story of true and lasting innovation fostered by interdisciplinary cooperation in the service of advancing science, a radical change that opens up new horizons in drug discovery and structural biology. In the future, the influence of DynamicBind is expected to spread its wings across the scientific domain, instigating more valuable revelations instrumental in enhancing healthcare results.
Article Source: Reference Paper | DynamicBind’s demo, instructions, and codes are available on GitHub, and the web server is available at https://neurosnap.ai/service/DynamicBind.
Follow Us!
Learn More:




Anshika is a consulting scientific writing intern at CBIRT with a strong passion for drug discovery and design. Currently pursuing a BTech in Biotechnology, she endeavors to unite her proficiency in technology with her biological aspirations. Anshika is deeply interested in structural bioinformatics and computational biology. She is committed to simplifying complex scientific concepts, ensuring they are understandable to a wide range of audiences through her writing.











{kind=link}