
In a groundbreaking study published in Nature Methods, a group of scientists consisting of the UC Santa Cruz Genomics Institute, University of Ferrara, Google LLC, and others individuated a new way of building customized pangenome references. The researchers made a great step forward in genomic research as they managed to overcome certain limitations of the reference genome strategy.
Understanding Pangenomes
Pangenome represents all the known genetic data for all the individuals within a species and not an average of a few sources. In other terms, it is a term used to grasp not just a reference genome of one or a few individuals but rather an entire population of various individuals that does not conform to the reference genome.
Why are Pangenomes important?
Reduces Reference Bias: Conventional reference genomes while conducting genomic data analysis are somewhat limiting since they tend not to capture the full picture of a genetic population. As pangenomes encompass greater amounts of genetic diversity, normalizing this bias becomes easier.
Improved Variant Calling: It also generates productivity in variant calling as, with pangenomes, it is easier to focus reads and find the genetic difference more efficiently without losing focus.
Enhanced Understanding of Genetic Variation: Pangenomes provide more details on the presence of genetic variation within a species, such as the localization of genes, the occurrence of specific alleles, and the genetic epidemiology of phenotypic traits.
Applications in Medicine and Biology: Pangenomes have a multitude of uses in medicine and biology, including detecting mutations that cause diseases, examining the evolutionary path of populations, or constructing individual therapeutics.
The need for personalized Pangenomes
Despite the merits provided by pangenomes, challenges still limit their full utilization. One of the biggest problems is the presence of unnecessary variants in the pangenome of an individual that is not part of the sample sequence being interrogated. These unnecessary variants will create false read mappings and unfortunately affect accurate genomic analysis.
To overcome this problem, scientists have come up with personalized pangenomes. As much as all these are termed pangenomes, personalized pangenomes are those that include only relevant genetic variations to an individual or group of individuals rather than the whole genetically diverse population.
Key Features of the Personalized Pangenome Approach
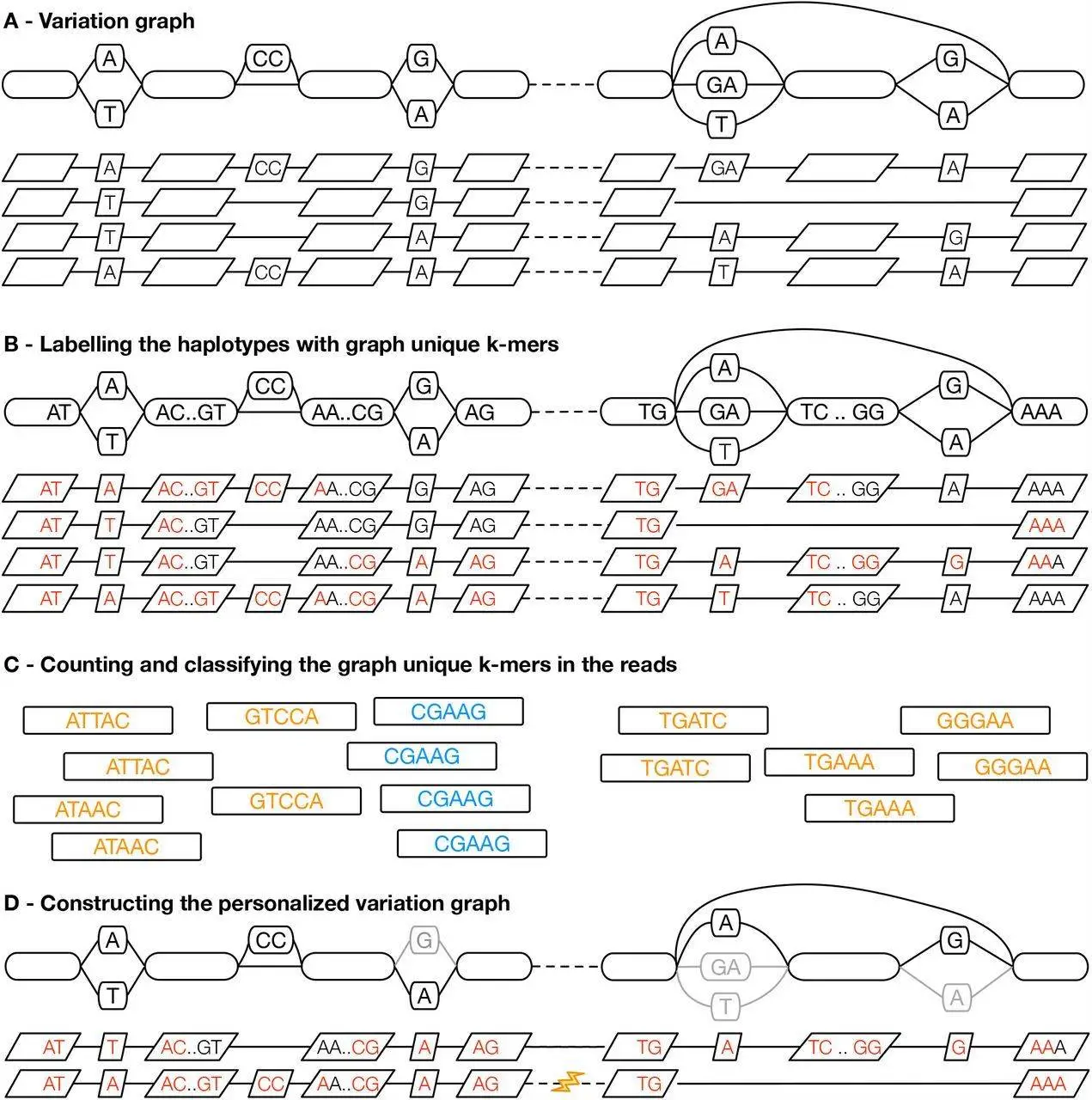
Haplotype Sampling: The procedure extracts local block haplotypes from the pangenome that are closely related in terms of genomic sequences. This guarantees the availability of the necessary allele variation in the personalized subgraph.
K-mer Based Analysis: K-mer counts in the reads are applied in k-mers classification and haplotype selection, hence a quick and effective strategy.
Integration with Giraffe Aligner: The constructed pangenome references are specially oriented to be used with the Giraffe aligner, greatly improving the mapping of short reads.
Creating Personalized Pangenome References: A Step-by-Step Guide
The personalized pangenome approach as described by researchers comprises the following critical stages:
Preprocessing Haplotypes:
- There is a preprocessing of the pangenome to obtain haplotypes and encode them appropriately.
- Haplotypes are structured in blocks to ease the process of sampling.
- For every block, graph unique k-mers are found that will be used for haplotype definition.
Counting K-mers in Reads:
- From the reads, k-mers are enumerated and classified as present, likely heterozygous, likely homozygous, or absent based on their counts.
- Excessively abundant K-mers are disregarded.
Sampling Haplotypes:
- The sampling of the haplotypes is done in a fashion in which the block is determined by the similarity to the sequenced genome.
- The highest-scoring haplotypes are selected using a greedy approach without restriction to homozygous or heterozygous Kmer counts.
- Full-length haplotypes are reconstructed from individual haplotypes that have been sampled.
Building the Personalized Pangenome Subgraph:
- A GBWT index is constructed for the purpose of archiving the haplotypes that have been sampled.
- A GBZ graph is developed from the haplotypes present in the index to form a personalized pangenome subgraph.
Results and Implications
The researchers evaluated their method using the Human Pangenome Reference Consortium (HPRC) graphs and real sequencing data. They found that in terms of accuracy and efficiency, personalized pangenome references fared far better than the conventional methods. In particular, this personalized schema led to a 74% decline in small variant genotyping errors compared to the Genome Analysis Toolkit (GATK) best-practice pipeline. In addition, it performed as well as long-read methods in genotyping structural variants.
Conclusions
The current study marks the evolution of genome analysis in the creation of personal pangenome references. By addressing the limitations of traditional reference genomes, the study will focus on dealing with the existing difficulties to make the process of investigating genetic variation simpler. This research is relevant in medical genetics, population genomics, and pharmacogenetics differences. With an increase in the size of the pangenome, as well as complexity, the more important will be the use of personal pangenome references—being able to extract much more out of genomic information.
FAQs
The construction of personalized pangenomes tends towards the medicine-aspected approach, which is towards selecting the relevant haplotypes from a global pangenome with the aid of k-mer counts in the reads. Mostly, this step can be defined as k-mer identification, where k-mers are present in the sample, and a haplotype containing such k-mers is selected.
Despite the numerous tasks in which personalized pangenomes can be applied, there are concerns about their application potential regarding the specific application. For instance, in the analysis of diseases due to their complex nature, pangenomes could be specifically suitable for polymorphic diseases.
The major concerns about its usage are the cost and its creation or usage of the personalized pangenome. Besides, there are instances when pangenomes do not perform well, like certain samples with low coverage or low frequency, and specific classes of mutation may not enjoy the benefits.
Personalized pangenomes are a field that is still in the rudimentary stages, and it has a bright outlook. It can therefore be expected that with the increase in the size and complexity of pangenomes, their personalization will be more valuable in genomic analyses.
Do you have any questions or comments about personalized pangenome references?
Please leave your comments below. We look forward to hearing from you!
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.





{kind=link}