
AlphaFold is a database of protein structures and predictions. It contains predictions for ~214 million proteins. AlphaFold2, developed by DeepMind, has dramatically scaled our capability to predict protein structures in silico. The extent of this data is overwhelming, and there are challenges regarding protein annotation, which practically applies to the information. Hence, researchers from the University of Trieste, SISSA (International School for Advanced Studies), ICTP (International Centre for Theoretical Physics), and IRCCS San Raffaele Institute introduced DPCstruct, an unsupervised clustering algorithm in the classification of protein domains using structural predictions informed by AlphaFold2. They discussed what DPCstruct means and what that might mean for the future of proteomics.
AlphaFold Database: A Revolution in Proteomics
As mentioned above, the AlphaFold database is a paranormal resource with structural predictions for almost 214 million proteins across a large diversity of species. We can explore protein structures as sequence data, too. This correctness in structural predictions could be very productively used in a large number of processes such as drug discovery, variant effect prediction, and understanding evolution at the level of the proteins. While great possibilities can indeed be achieved using AlphaFoldDB, the lack of proper descriptions of full proteins greatly reduces their usability and demands some advanced tools for the classification and annotation of such structures.
Protein Annotation Challenge:
Protein annotation means ascribing functional information to either protein sequences or structures, a necessary task to make sense of the huge data arising from AlphaFold2. Conventionally, protein annotation has been carried out using approaches based on sequences, which depend on homology for predicting functions. These methods, however, have significant limitations, especially when applied to distant homologs, where proteins may have low sequence similarities but similar structures. Consequently, a large number of protein databases remain uncharacterized. These limitations can be addressed through structure-based predictions, which find remote homologs and thus give more accurate predictions of protein functions.
Introducing DPCstruct: A Novel Clustering Algorithm
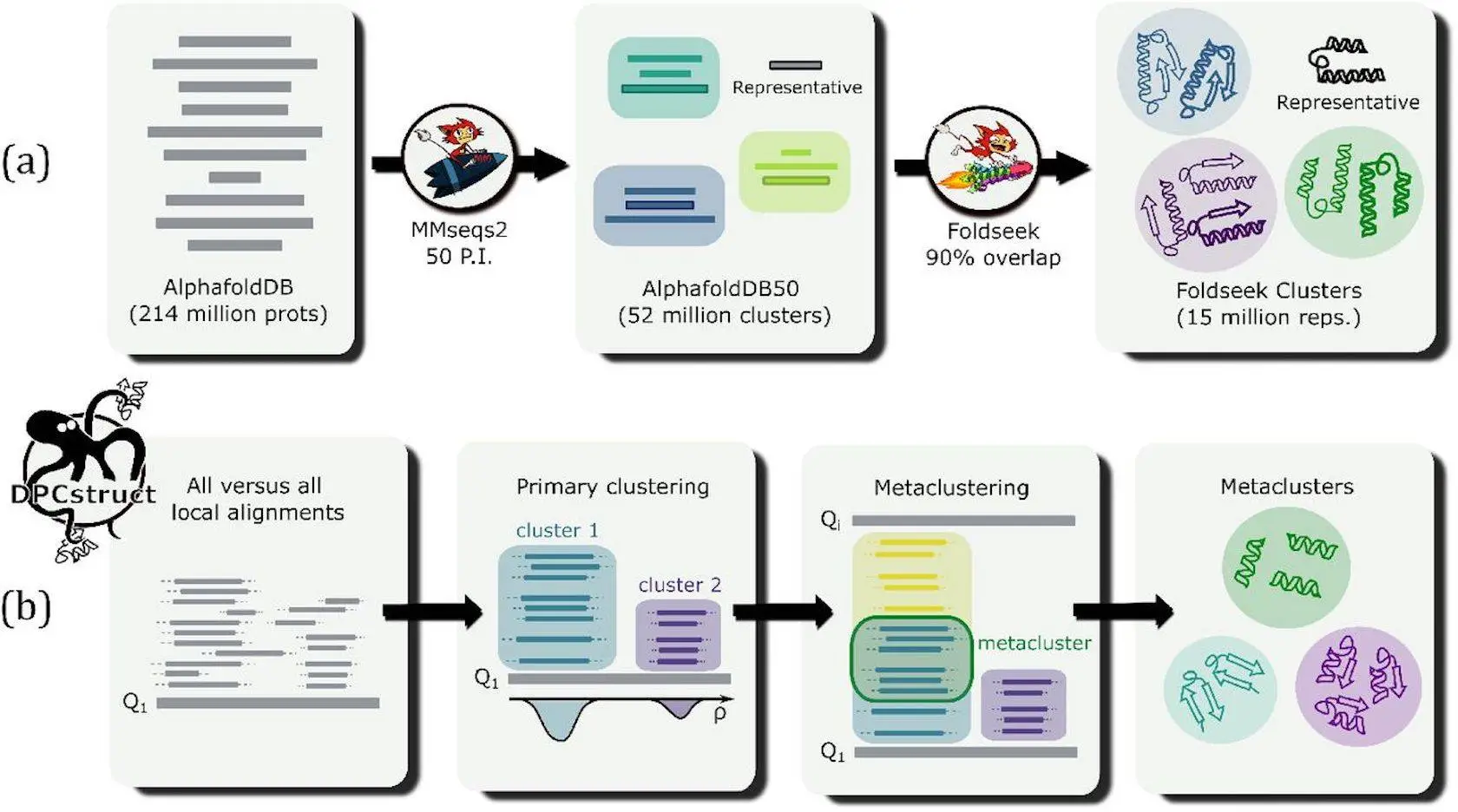
DPCstruct stands for Domain-Protein Clustering using Structural predictions. It is a new unsupervised clustering algorithm that classifies protein domains using structural predictions emanating from AlphaFold2. This is the idea behind the assumption that structural similarity will be a better estimator of functional similarity than sequence similarity alone. DPCstruct uses all-against-all local alignments given by Foldseek-a, a fast and sensitive structural aligner-detected in order to find and cluster very frequently recurring structural motifs into what we call “domain clusters.” These clusters represent structurally similar regions of proteins or domains, which can give some hint about the function and evolutionary relation of the proteins.
How does DPCstruct Work?
DPCstruct works in several steps:
- First, it executes all-against-all Foldseek with the input database, producing local structural alignments.
- Then, these alignments are grouped into primary clusters using a density-based clustering algorithm, representing putative domains.
- Lastly, the primary clusters merge to form meta-clusters, each comprising structurally related protein domains. Thus, such an approach provides an all-encompassing classification of protein domains that captures both known and novel structural relationships.
Benchmarking DPCstruct: Performance and Results
DPCstruct was run on the Foldseek Cluster database, a representative set of proteins for Alpha-FoldDB. The algorithm detected 28,246 metaclusters, and they compared membership sets of these metaclusters to the established databases of protein folds, namely SCOP and CATH. It was shown that DPCstruct can recover most protein folds cataloged in the databases SCOP and CATH. More precisely, DPCstruct achieves a 94% recall in the set of SCOP folds, while CATH folds achieve 86%. The algorithm also identified various new clusters of proteins, and 24% of the metaclusters did not show similarities either structurally or sequentially with any of the known protein families, therefore suggesting the fact that DPCstruct was able to identify novel protein domains and folds-which are usually unexplored.
Consistency with Sequence-Based Databases
In another assessment, protein family DPCstruct results were compared with the Pfam database- a major resource for protein family classification based on the sequence-similarity principle. This time, elements were compared for consistency between DPCstruct domain classifications and Pfam clan-level annotations. The analysis showed that 70% of DPCstruct metaclusters were fully consistent with Pfam annotations, pointing to a strong level of agreement between the structural and sequence-based classifications. However, Pfam misclassifications were also suggested by DPCstruct, further highlighting the fact that it had disclosed new structural relationships that could not have been detected by methods based on sequence only.
Novel Protein Domain Discovery
The identification of unknown metaclusters stood out as one of the most important results from the analysis, which represented putative novel protein domains. These metaclusters represent 24% of the total number of identified, and yet there was no resemblance to any structural similarity of known protein families or folds found in established databases. Indeed, the identification of such new domains illustrates an emerging potential role that DPCstruct may very soon play in expanding our knowledge of the protein universe! Isn’t that great? It also offers new targets for functional investigations and can help augment our understanding of protein structure and function.
Conclusion
DPCstruct is a big advance in proteomics as it offers a scalable and efficient solution for the classification of protein domains based on structural predictions. DPCstruct, which combines the great power of accurate structural data provision by AlphaFold2 with full alignment and template-wise search capabilities of Foldseek, has advanced the ability to annotate the vast protein space. This property makes the enzyme an ideal candidate for researchers in the field of proteomics, including metagenomics, all of which are part of large-scale studies. In this way, tools such as DPCstruct, among others, will contribute to taking full advantage of the protein data through the golden era that structural biology is undergoing, leading to great new applications and revelations in the biological and medical sciences.
Article Source: Reference Paper | The DPCstruct algorithm is available on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Neermita Bhattacharya is a consulting Scientific Content Writing Intern at CBIRT. She is pursuing B.Tech in computer science from IIT Jodhpur. She has a niche interest in the amalgamation of biological concepts and computer science and wishes to pursue higher studies in related fields. She has quite a bunch of hobbies- swimming, dancing ballet, playing the violin, guitar, ukulele, singing, drawing and painting, reading novels, playing indie videogames and writing short stories. She is excited to delve deeper into the fields of bioinformatics, genetics and computational biology and possibly help the world through research!











{kind=link}