
Genes are the basic units of heredity, but they only come to life when translated into proteins. The study of the proteins found in cells and tissues is called proteomics and is largely dependent on Mass Spectroscopy (MS) techniques. Such MS-based proteomics datasets are on the rise, requiring advanced tools to analyze them and uncover new biological discoveries. Jayantha Gunaratne’s team of researchers from A*STAR, Singapore, describes the use of advanced machine learning techniques to extract novel biomarkers from proteomics data, specifically focusing on finding effective protein markers for high-grade serous ovarian carcinoma (HGSOC).
Why is HGSOC important?
High-Grade Serous Ovarian Cancer is a highly malignant form of ovarian cancer characterized by late-stage diagnosis and poor prognosis. It comprises high mortality rates with a five-year survival rate of 34%. Accurate detection requires biopsies, which are invasive and could lead to several complications. Current approaches for detecting High-Grade Serous Ovarian Cancer (HGSOC) rely on serum biomarkers like CA125 and HE4, which are generally used for ovarian cancer detection. However, these methods lack specificity for HGSOC. Hence, we require non-invasive methods such as biomarkers to safely and accurately diagnose HGSOC patients.
Data Acquisition
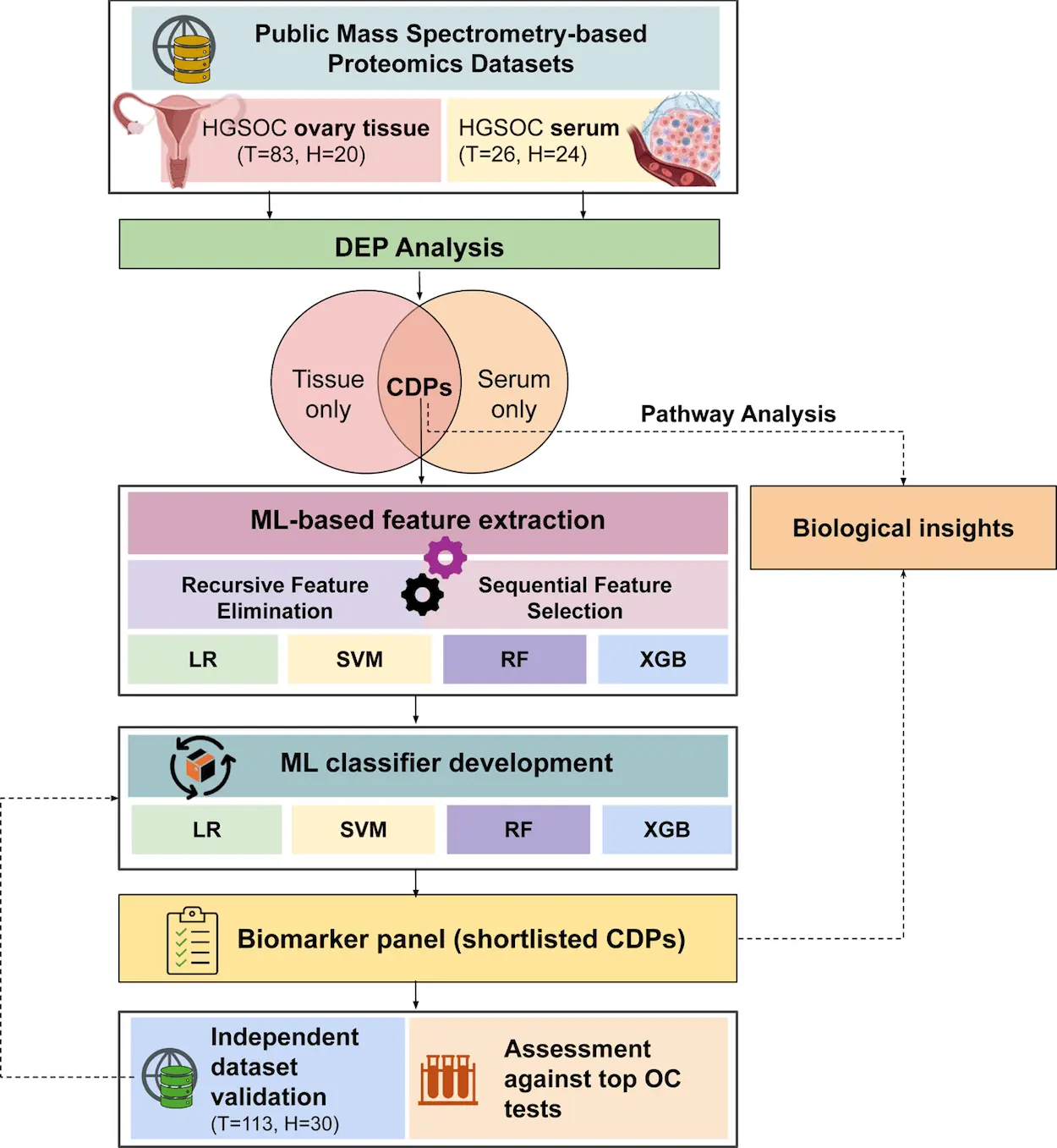
The study utilized publicly available proteomic datasets from both ovary tissue and serum samples consisting of 153 participants. There were 109 HGSOC patients and 44 healthy controls. The tissue dataset included label-based proteomic data, while the serum dataset was label-free. Below, we explain the Machine Learning pipeline developed by the researchers.
The Machine Learning-Based Biomarker Extraction Pipeline
Protein biomarkers are specific types of biomarkers that involve the measurement of proteins. Certain proteins must play a significant role in HGSOC. We require their biomarkers for proper analysis and prediction. The researchers first looked at proteins that were co-dysregulated, meaning they showed similar patterns of abnormal expression among a group of proteins that were differentially expressed in both tissue and serum samples. They used specific statistical criteria to identify these co-dysregulated proteins (CDPs) in the datasets. In total, they found 88 CDPs in both datasets, indicating that these proteins may play a significant role in HGSOC.
Next, to identify their discriminative biomarkers, an ML exercise was applied, which included feature selection and classifier development.
Feature Selection
Recursive Feature Selection
Recursive Feature Selection (RFS) iteratively trains a model and removes the least important features, re-evaluating the model after each removal until a stopping criterion is met. This method was applied to CDPs belonging to clusters 1 and 2 using 20% of patient samples from the serum dataset.
Sequential Feature Selection
Feature Selection (SFS) adds or removes features depending on how they contribute to the model’s performance. This method was also applied to CDPs belonging to clusters 1 and 2. Choosing the classifiers was the next task.
Classification using Machine Learning Models
Machine learning models like Logistic Regression (LR), Support Vector Machines (SVM), Random Forests (RF), and Extreme Gradient Boosting (XGB) were used with 5-fold cross-validation to select serum markers for distinguishing between HGSOC and healthy individuals. Recursive Feature Selection identified 10 serum markers from CDP cluster 1 and 32 markers from cluster 2 based on the cross-validation accuracy. Sequential Feature Selection was then applied using the F1 score to determine the optimal number of biomarkers, leading to the identification of EEF1G, MSLN, BCAM, and TAGLN2 for cluster 1 (0.97 AUC) and CRISP3 and MMP9 for cluster 2 (0.98 AUC) as the most distinctive markers.
Two ML models were created using 60% of patient samples from the serum dataset to assess the performance of the markers. These markers were utilized as features in these ML models. Four different ML classifiers were compared using those markers to determine the most suitable classifier for the task. After comparing the performance of the ML classifiers, the XGB models were selected as the final classifiers. Researchers next quantified the performance of the new biomarkers, suggesting HGSOC prevalence.
Assessing Performances of New Biomarkers
Researchers tested the performance of the identified biomarkers using two publicly available datasets. Ten HGSOC patients and ten healthy people made up the serum dataset. The markers, EEF1G, MSLN, BCAM, and TAGLN2, accurately identified HGSOC samples with an Area Under the Curve of 93% and an F-score of 92%. The CRISP3 and MMP9 marker pair had an AUC of 83% and an F1 score of 86%.
Conclusion
Detecting HGSOC was accomplished cleverly by using proteomics datasets that were influenced by mass spectroscopy techniques. The combination of classical ML techniques with proteomics data analysis has led to the identification of novel biomarkers with high accuracy in diagnosis. Through these findings, researchers can aim to improve early detection and treatment outcomes for HGSOC patients and possibly save countless lives.
Article Source: Reference Paper | Code available on – GitHub.
Follow Us!
Learn More:




Neermita Bhattacharya is a consulting Scientific Content Writing Intern at CBIRT. She is pursuing B.Tech in computer science from IIT Jodhpur. She has a niche interest in the amalgamation of biological concepts and computer science and wishes to pursue higher studies in related fields. She has quite a bunch of hobbies- swimming, dancing ballet, playing the violin, guitar, ukulele, singing, drawing and painting, reading novels, playing indie videogames and writing short stories. She is excited to delve deeper into the fields of bioinformatics, genetics and computational biology and possibly help the world through research!











{kind=link}
[…] Modeling the Perfect Diagnosis with Machine Learning in Ovarian Cancer […]