

Researchers at Mellon University and MIT introduce VibeGen, a generative AI framework for end-to-end de novo protein design conditioned on dynamics. The model generates de novo proteins that not only fold but also exhibit the required dynamics.
Proteins and biomolecular structures are the backbones of many functions in our bodies. They catalyze reactions, transmit signals, and provide structure to our skin and hair. By designing new proteins, scientists can create new medicines, develop synthetic biology tools, and even solve industrial problems.
But proteins are not static; they are constantly moving and shifting. These movements are either ultra-fast vibrations (at the atomic level) or slower rearrangements, like the whole domains shifting. Markus J. Buehler, the co-author of the research, often describes proteins as ‘nature’s molecular machines of motion’ and stresses that designing for dynamics can unblock adaptive biomaterials and flexible enzymes, in specific cases such as p53 and CFTR mutants, where dynamics tend to go wrong.
Designing or Engineering Proteins the Traditional Way
Previous techniques like NMR spectroscopy, Cryo-EM, smFRET, etc., helped us in mapping timescales and amplitudes of changes in proteins. By combining these experimental insights with computational models, researchers could decode how the motion of proteins drives functions.
Other computational methods that were powerful, like MD simulations, Normal Mode Analysis (NMA), and Elastic Network Models (ENM), were also too expensive, and scaling to thousands of proteins was impossible. That’s why efficient generative approaches are needed.
VibeGen is built on this foundation; instead of only predicting static structures, it designs proteins conditioned on vibrational modes directly informed by kinds of motions NMR, smFRET, and tetrahertz spectroscopy reveal.
Current State of Protein Design with AI: Why VibeGen Is a Step Forward
Deep learning and generative AI have revolutionized protein research. Tools like AlphaFold2 and RoseTTAFold can now take a protein’s sequence and predict its 3D structure with accuracy close to experiments, just faster and cheaper. This breakthrough allowed us to study proteins at the orphan sequence to protein complex levels. However, most folding tools focus on static structures, ignoring dynamics, missing out on a huge part of how proteins actually work.
VibeGen is one of the first frameworks to tackle this head-on; it doesn’t just predict structures, but it designs proteins conditioned on dynamics. VibeGen integrates vibrational modes into the generative process, making sure that proteins are designed not just to fold but to move in specific ways.
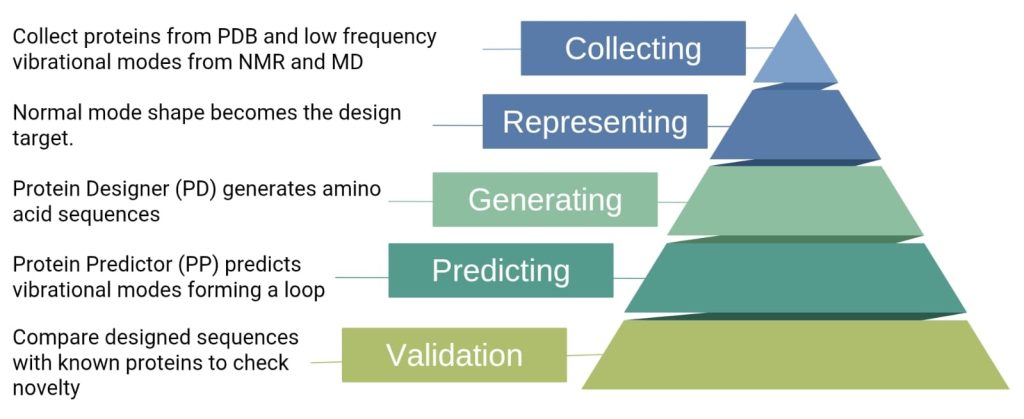
Dual Model Architecture: The VibeGen Workflow
The two agents, namely protein designer (PD) (ESM-2, 150M parameters) and protein predictor (PP) (U-Net with attention), are built as separately trained protein language diffusion models (pLDMs). A pretrained protein language model (pLM) is already trained on a huge protein sequence dataset, and the diffusion model is the trainable part that learns to generate or denoise sequences conditioned on dynamics.
PD and PP work together as a collaborative loop. The predictor proposes amino acid sequences that should produce a desired vibrational mode, while PP predicts the vibrational mode for a given sequence, checking if it matches the target.
Key Results and Implications
Research proved that the model could learn the complex degenerate relationships between protein sequences and their vibrational modes:
Accuracy of designs: The model managed to successfully generate proteins whose normal mode vibrations matched the input design. Quantitatively, Pearson’s correlation coefficients between target and measured profiles peaked around 0.87 (with a median of ~0.53), showing a good accuracy at the overall shape level, though residue level precision still stands as a challenge. Examples include L-shaped, U-shaped, W-shaped, and multi-peak vibrations.
Novelty and structural features of sequences: Even when trained on existing PDB proteins, the system produced sequences that diverged from natural ones, generating many de novo proteins. These proteins folded into stable structures predicted by OmegaFold.
Dual-model synergy: The two-agent system outperformed single-model baselines, amplifying forward prediction and inverse design even while balancing diversity, accuracy, and novelty better than a single end- to-end model.
Future Directions
- Researchers’ current work focuses on the lowest non-trivial normal mode. Future models could incorporate multiple low-frequency modes.
- The PD-PP duo could be extended into larger multi-agent systems where different agents specialize in dynamics, binding, stability, etc.
- A move towards fully automated workflows integrating dynamics with other design goals will allow multimodal design frameworks.
- Researchers are also planning to apply the model to larger proteins and complexes and introduce screening strategies to ensure more de novo sequences with more accuracy.
Article Source: Reference Paper arXiv | Published Abstract | Reference Article
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Saniya is a graduating Chemistry student at Amity University Mumbai with a strong interest in computational chemistry, cheminformatics, and AI/ML applications in healthcare. She aspires to pursue a career as a researcher, computational chemist, or AI/ML engineer. Through her writing, she aims to make complex scientific concepts accessible to a broad audience and support informed decision-making in healthcare.











{kind=link}