
Researchers at the University of Washington, USA, have developed a new AI system called RoseTTAFoldNA that can rapidly and accurately predict the 3D structures of proteins bound to DNA and RNA. The ability to model these protein-nucleic acid complexes has important implications across fields from biomedicine to biotechnology. RoseTTAFoldNA expands on the award-winning protein structure prediction system RoseTTAFold. It can generate high-confidence predictions for complexes with error rates considerably lower than previous state-of-the-art techniques. This advance should provide an invaluable resource for understanding natural protein-DNA and protein-RNA interactions and designing novel nucleic acid targeting proteins.
Development of RoseTTAFoldNA
Protein-nucleic acid complexes play an essential role in biology as by studying their stereochemistry, it is possible to comprehend different cellular processes like gene expression, replication, transcription, translation, cell metabolism, and enzymatic events. The current methods for predicting the structure of protein-nucleic acid complexes comprise modeling the individual components of the proteins and nucleic acids and then utilizing computational docking calculations to assemble the complexes.
In most cases, RNA structure prediction starts with the secondary structure (Watson/Crick base pairing) prediction and subsequently involves building the secondary structure into a tertiary structure. More recently, models have been chosen from ensembles created using alternative structure sampling techniques, and contact prediction has been employed to help determine RNA structure. The ability to predict the structures of nucleic acids and protein-nucleic acid complexes has advanced, but it still lags far behind the ability to predict the structures of proteins based on their amino acid sequences. High-accuracy methods like AlphaFold and RoseTTAFold solve this problem.
One or more aligned protein sequences are sent into AlphaFold and RoseTTAFold, which then process the data in parallel to create 1D, 2D, and, in RoseTTAFold’s instance, 3D tracks before producing three-dimensional protein structures. These deep networks learn their 10s to 100s of millions of free parameters by training on massive protein datasets with known structures from the PDB.
RoseTTAFold and AlphaFold2 can produce precise models of protein complexes as well as monomers by simulating folding and binding over hundreds of cycles of subsequent transformations. RoseTTAFold has been extended to predict nucleic acid structures and protein-nucleic acid complexes using PDB structures. This work expands on the end-to-end deep learning methodology, acquiring new parameters for generic protein-nucleic acid systems and enhancing the prediction of nucleic acids.
RoseTTAFoldNA Enabling Precise Protein-Nucleic Acid Complex Prediction
RoseTTAFoldNA is a single trained network that generates 3D structure models quickly with confidence estimates for RNA tertiary structures, protein-DNA, and protein-RNA complexes. Confident predictions are significantly more accurate than state-of-the-art techniques in each of the three scenarios. RoseTTAFoldNA is expected to be broadly helpful in the design of sequence-specific RNA and DNA binding proteins as well as in simulating the structure of naturally occurring protein-nucleic acid complexes.
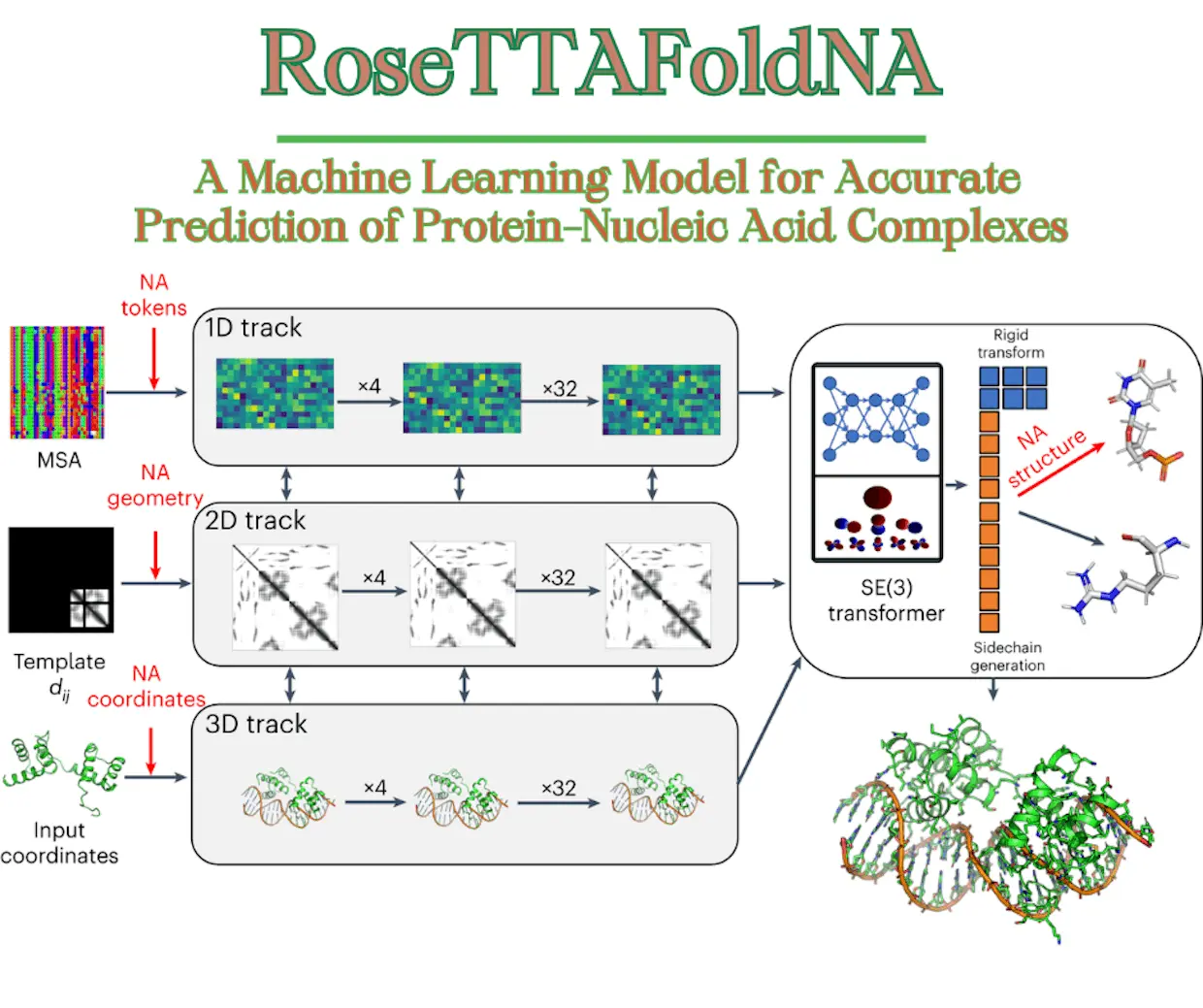
RoseTTAFoldNA’s architecture is derived from RoseTTAFold’s three-track design, which refines three biomolecular system representations simultaneously: sequence (1D), residue-pair distances (2D), and cartesian coordinates (3D). Researchers expanded all three of the network’s tracks to accommodate nucleic acids in addition to proteins and also made a number of performance-enhancing changes.
1D track of RoseTTAFoldNA: 22 tokens, corresponding to the 20 amino acids, a 21st “unknown” amino acid or gap token, and a 22nd mask token that enables protein design+10 additional tokens, corresponding to the 4 DNA nucleotides, the 4 RNA nucleotides, unknown DNA, and unknown RNA.
2D track of RoseTTAFoldNA: a representation of the interactions between all pairs of amino acids in a protein or protein assembly;+ generalized the 2D track to model interactions between nucleic acid bases and between bases and amino acids.
3D track of RoseTTAFoldNA: represents the position and orientation of each amino acid in a frame defined by three backbone atoms (N, CA, C) and up to four chi angles to build up the sidechain.+ representations of every nucleotide with ten torsion angles that let the nucleotide assemble into its entirety and a coordinate frame that describes the location and orientation of the phosphate group. With 67 million parameters, RoseTTAFoldNA is made up of 36 of these three-track layers and four more levels for structural refinement.
The study trained an end-to-end protein/NA structure prediction network using a combination of protein monomers, protein complexes, RNA monomers, RNA dimers, protein-RNA complexes, and protein-DNA complexes. Sequence similarity searches were used to produce multiple sequence alignments (MSAs) of related protein and nucleic acid molecules. Network parameters were optimized by minimizing a loss function and assessing error prediction accuracy, recovery of masked sequence segments, and residue-residue interaction geometry. To reduce redundancy, physical information was included in the loss function during fine-tuning and as input features in the final refinement layers. Ten percent of the clusters were held back for model validation during training.
The validation set used RNA and protein/NA structures solved after May 2020, with complexes handled as complete complexes. Paired MSAs were produced for complexes, including multiple protein chains. GPU memory limitations excluded cases containing multiple protein chains, RNA chains, or DNA duplexes, while 41 cases had a single RNA chain, 21 cases had one protein molecule, and 67 cases had multiple DNA duplexes.
Performance and Application
The findings indicate that the available data is adequate for de novo structure modeling in numerous instances, with the accuracy of RNA prediction being almost equal to that of protein structure prediction. Approximately 31% of the cases involve accurate modeling of protein/NA interfaces without the need for shared Multiple Sequence Alignment (MSA) information or homologs with known structures. Blind and prospective testing will be crucial for a more thorough, critical assessment of the procedure.
The predictive capabilities of RoseTTAFoldNA for protein-nucleic acid complexes are quite encouraging. At greater confidence levels, the accuracy of the model increases. The average lDDT (Local Distance Difference Test) for all forecasts is 0.73, and the average lDDT for predictions with extremely high confidence (plDDT > 0.9) is 0.84. While structures with close homologs in the training set tend to yield more accurate predictions, numerous structures without any observable homologs in the training set also yield correct predictions. The number of sequences in the MSA increases the accuracy, yet many single-sequence cases are predicted correctly.
In RoseTTAFoldNA, confident predictions have considerably higher accuracy than current state-of-the-art methods. This model will not only apply to complexes composed of one type of protein subunit but can be used universally in modeling the structures of naturally occurring DNA and RNA-binding proteins and even designing RNA or DNA–binding proteins.
Conclusion
A significant advancement in the prediction of protein–nucleic acid complex structure is demonstrated by RoseTTAfoldNA. Because of this, the interest in studying the phenomenon of proteins binding to particular acids and nuclear sequences will grow, and in the future, scientists may find great use in this model. As the need for precise resolution for such structures grows, the significance of models like RosettaFoldNA in expanding our understanding of biological processes involving protein-nucleic acid complexes cannot be emphasized more.
Article Source: Reference Paper | RoseTTAfoldNA code is available on GitHub
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}