
Significant progress has recently been made in predicting protein sequence to structure; however, modeling protein interfaces and complex structures is still tricky, particularly when binding partners undergo large conformational changes. According to earlier research, in only 43% of cases, AF-multimer (AFm) can accurately predict protein complexes. In this work, Johns Hopkins University researchers combine a replica exchange docking algorithm based on physics with AlphaFold, a structural template generator. The effectiveness of AlphaFold confidence measures (pLDDT) in predicting protein flexibility and docking accuracy for multimers has been demonstrated using a carefully selected set of 254 protein targets with both unbound and bound structures. The ReplicaDock 2.0 protocol, which incorporates metrics for precise protein complex structure prediction, makes this possible. For 66% of benchmark targets, AlphaRED (AlphaFold-initiated Replica Exchange Docking) produces CAPRI forecasts of an acceptable grade or higher after docking unsuccessful AF predictions. This approach blends physics-based increased sampling with deep-learning architectures trained on evolutionary data.
Introduction
Deep learning techniques combined with evolutionary data are being revolutionized in the field of structure prediction by structural biology. AlphaFold, a program that has demonstrated excellent single-chain structure prediction accuracy, has been used for the prediction of protein complexes. Since conformational changes generated by binding are a common feature of protein-protein interactions, it is important to understand their dynamics. The accuracy of energy functions, sampling duration, and length scales have been constraints for computational methods that have tried to sample the unknown terrain of protein-protein interactions. Just 43 percent of the time, AlphaFold-multimer (AFm) can accurately predict protein complexes. The prediction of protein assembly and binding routes may be strengthened by adding a biophysical context as DL-based methods develop.
About AlphaFold
AlphaFold is a powerful tool for protein docking that was developed as a result of recent developments in structural biology. This method blends traditional physics-based sampling with deep learning tools to extract optimal results. AlphaFold helps reduce sampling degrees of freedom by offering a snapshot of pertinent local minima. Understanding protein dynamics, adjusting protein-protein interactions, and using this knowledge to improve protein design are all made possible by the incorporation of physics into deep learning models.
Looking into the Features of AlphaFold-multimer with Physics-based Docking Schemes
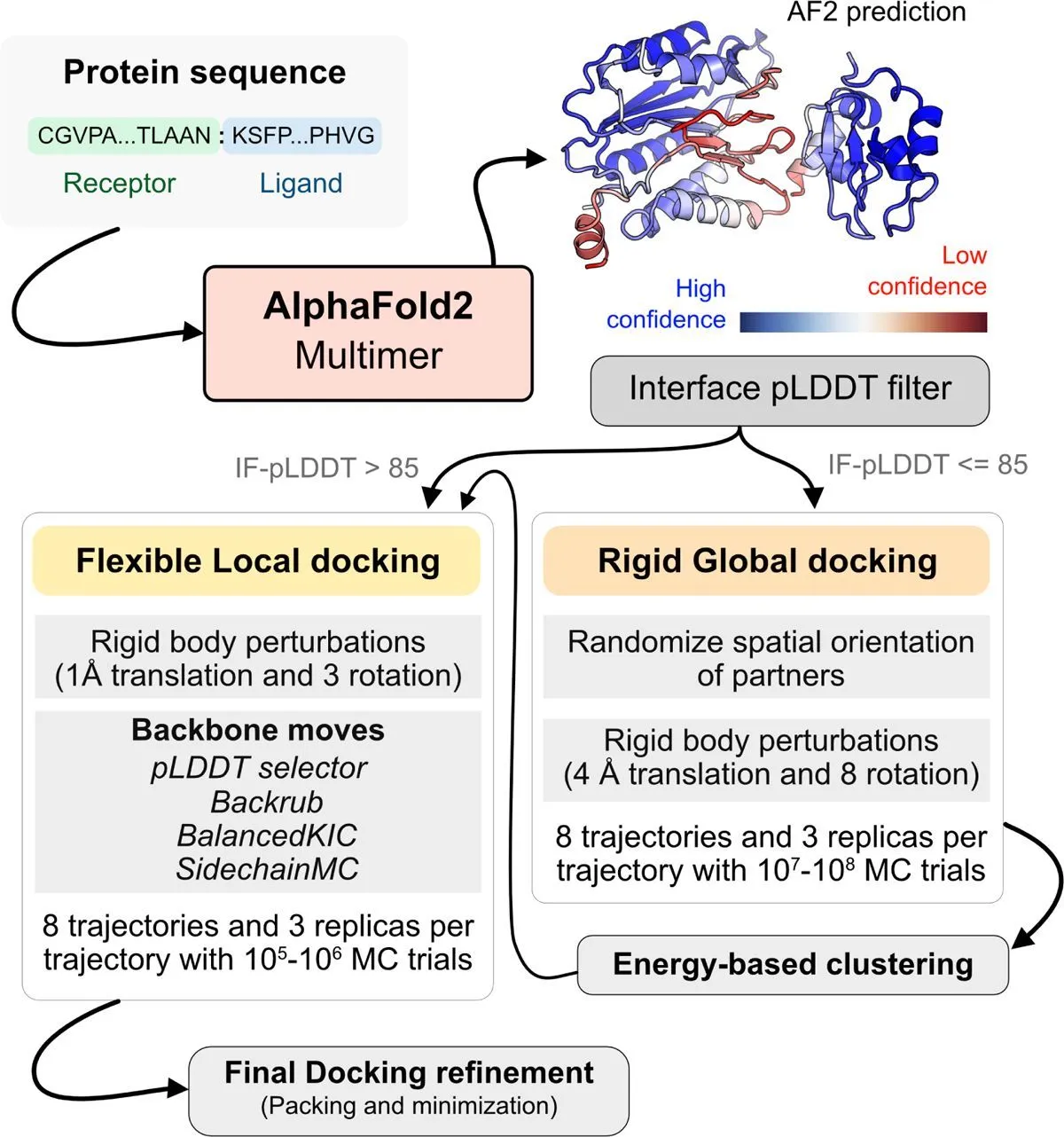
An efficient pipeline for modeling protein complexes is created by combining deep learning AlphaFold and physics-based docking techniques. The objectives are to resolve AFm failures and capture conformational changes brought on by binding. The binding site confidence and conformational flexibility are detected using the AlphaFold-multimer method. ReplicaDock 2.0 receives the structural template produced by AFm, resulting in the pipeline known as AlphaRED. Benchmark targets of bound and unbound protein structures are used to evaluate the accuracy of AlphaRED. These targets include antibody-antigen interfaces, which present a challenge to AF2m because of the lack of evolutionary information across the interface. In order to anticipate complicated protein complexes, this method seeks to integrate the best features of deep learning with biophysical techniques.
Methodology
Initially, researchers demonstrated that docking accuracy and flexibility may be estimated using AlphaFold confidence measures. A reliable metric to assess the accuracy of AFm projected binding interfaces is interface-pLDDT, which is the average of the per-residue pLDDT just for the interfacial residues. Furthermore, per-residue pLDDT thresholds can identify regions of backbone flexibility at binding. As a result, ReplicaDock 2.0 can employ the anticipated models from AFm as input structures to help determine the mobile protein segments and make decisions about global versus local sampling. One can effectively explore the protein energy landscape using DL methods for downstream sampling with a physics-based energy function and structure prediction, as shown by AlphaRED’s performance on DB5.5.
In order to explore the applicability of the approach to blind protein targets, researchers lastly assessed the most recent CASP15 targets. AlphaFold’s training routine did not include CASP15 targets, which functioned as blind challenges to determine the protocol’s effectiveness. Using AlphaRED, researchers were able to achieve DockQ scores greater than 0.23 for each of the five targets. Targets T205, T207, and T208, in particular, all had medium-quality models (DockQ > 0.49). Stochastic perturbation with dropout and enhanced sampling were used by AFSample, a top-performing group in CASP15, to produce medium and high-quality models for these targets. To generate about 240x models with a compute time around 1000x more than the baseline AFm, the AFSample necessitates GPU simulations.
For the docking procedure using the baseline version, researchers generated 1–5 structures using ColabFold. Using the docking routine fused with ColabFold, they saved several days on GPU (since each GPU node can have up to 48 cores) and only needed 5-7 hours on the CPU cluster (which runs on a single node with 24 cores or approximately 100 hours of CPU-hours per target). Protein complex structure prediction may now be accomplished more accurately and within reasonable compute times thanks to the AlphaRED docking technique.
Conclusion
In structural biology, AlphaFold is a cutting-edge technique that uses sequences to predict protein structures, including those of monomers, complexes, and larger assemblies. Exploring the evolutionary connections among amino acids in different protein families enhances the precision of monomer predictions. Nonetheless, insufficient evolutionary constraints may result in imprecise binding locations throughout protein interfaces. In order to overcome this, AlphaFold’s predictions are enhanced using an energy-function dependent sampling technique, which results in improved backbone conformational diversity and precise protein complex structure prediction. For all targets, the AlphaRED approach reduces failure cases in AFm projected models; overall, 66% of targets produced CAPRI acceptable-quality or superior models. The precise design and optimization of antibody-antigen targeting have a significant impact on the success rate of antibody therapies. This work facilitates high-throughput computational screening by addressing the deficiency of quick docking techniques for antibodies. Refinement via integration of a physics-based approach might be able to manage non-canonical residue types or post-translationally altered proteins.
Article Source: Reference Paper | AlphaRED pipeline is available on GitHub | An online server implementation is available on the Gray lab ROSIE server.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}