
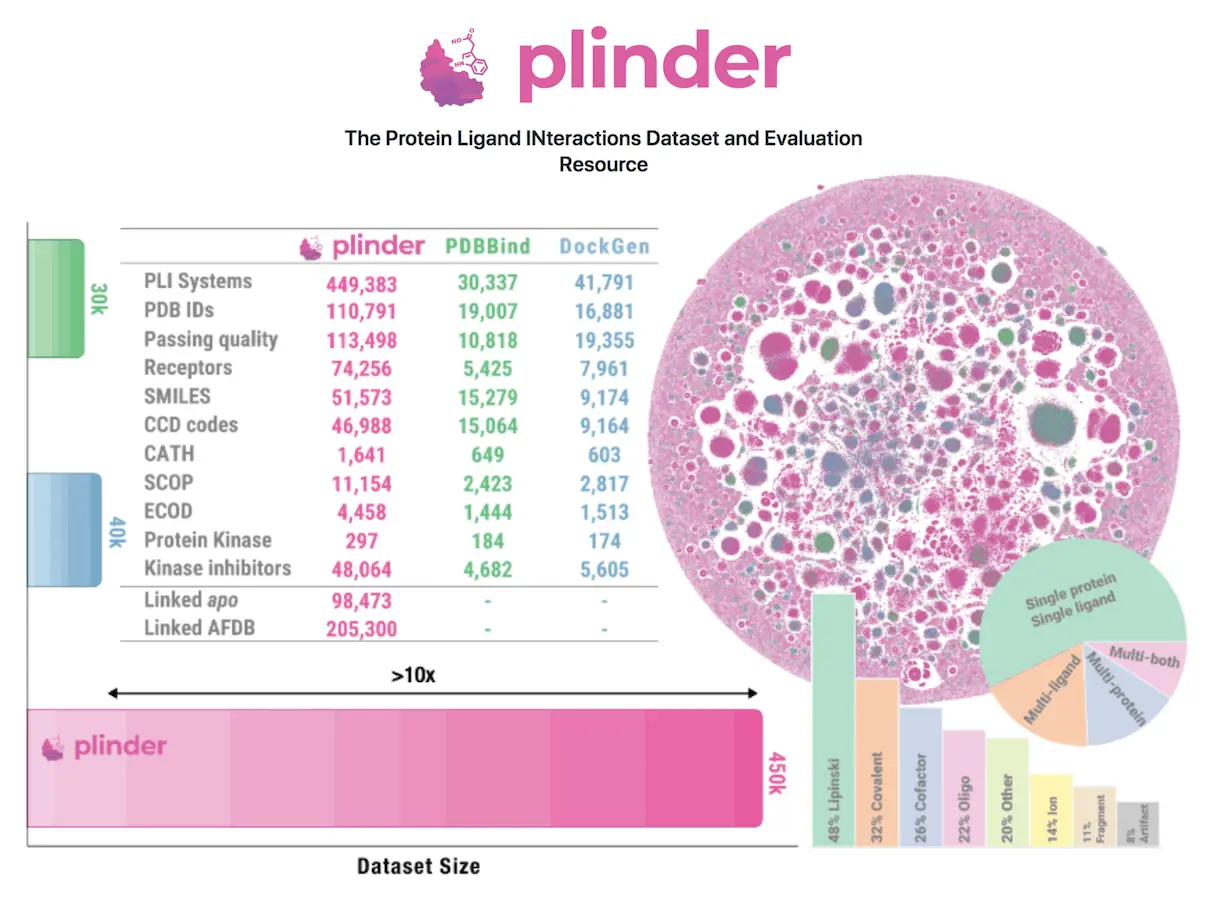
Small molecule drug design depends on protein-ligand interactions, and achieving experimental precision necessitates a wide and well-curated dataset. The scarcity of current datasets makes it difficult to realistically evaluate the generalization capabilities of methods and leads to the leakage of training data, underscoring the need for a diversified approach. PLINDER (protein ligand interactions dataset and evaluation resource), the largest and most annotated dataset to date, was presented by researchers to solve these inadequacies. It consists of 449,383 PLI systems, each with over 500 annotations, similarity metrics at the protein, pocket, interaction, and ligand levels, as well as matched unbound (apo) and projected structures.
Introduction
Protein structure and behavior predictions are made by the protein-ligand field using deep learning-based prediction techniques. This covers tasks such as co-folding, pocket-conditioned ligand synthesis, ligand-conditioned protein engineering, flexible pocket docking, stiff body docking, and molecular scaffolding. These techniques facilitate side-chain motions of pocket residues by predicting the position of a ligand within a particular protein structure. Moreover, ligand affinity for a protein or pocket is increased by molecular scaffolding.
Protein structures are essential for protein engineering and drug discovery, with precise ligand binding posture prediction being a critical component of these techniques. Nevertheless, the training and assessment datasets have an impact on how effective these techniques are. Realistic inference scenarios, test set diversity, test set quality, little information leakage between test and train, and diversity of training sets are important factors to consider. In addition to preventing over-fitting and demonstrating performance across a range of complicated types and use cases, these considerations aid in evaluating generalization capabilities. For protein engineering to be successful, several elements must be considered.
Protein-ligand interaction structural datasets must take several critical factors into account to be useful. An extensive dataset called BioLip2 places a strong emphasis on functional annotation, although it is not partitioned well enough for machine learning techniques. While PDBBind and its derivatives have proposed splits, they are modest and include information leakage. The datasets used in attempts to address leakage concerns in PDBBind are small, and there is no systematic assessment of various metrics. DockGen provides unique test domains by carefully merging datasets using evolutionary classification of protein domains (ECOD) annotations; nevertheless, it is restricted by biases in manual curation and dataset size.
Genesis of PLINDER
Protein-ligand complexes come in a variety of forms, including multi-ligand systems, oligonucleotides, peptides, and saccharides, and PLINDER provides the broadest and most varied dataset available. To quantify diversity and identify information leakage, it computes similarities between complexes at the protein, pocket, PLI, and ligand levels.
In addition, PLINDER suggests a method to prioritize a varied, excellent test set with little leakage and annotates complexes for quality and domain information. To provide plausible inference scenarios, it additionally connects holo complexes to pertinent apo and anticipated structures. By taking these steps, PLINDER hopes to give scientists studying proteins and ligands a solid and trustworthy dataset to use in the testing and training of deep learning-based prediction techniques, which will ultimately lead to the creation of new strategies for protein engineering and drug discovery.
Impact of PLINDER on Computational Biology
In Protein-Ligand Interactions – PLINDER has extracted 1,344,214 PLI systems from 162,978 PDB entries, of which 449,383 are holo systems. Of these systems, 34% meet the X-ray high-quality requirements, 26% have more than one ligand, and 25% have more than one interacting protein chain. A dataset of apo chains with no discernible ligand interactions is also offered by PLINDER, which is automatically curated. 37% of the 615,932 ligands in holo systems meet the Lipinski Ro5 requirements, and 23% have a covalent connection. PLINDER is a member of 2,117 congeneric MMS (matched molecular series), all of which have three or more ligands with a common core. The curation method allows for the simultaneous identification of apo chains and holo systems over the whole PDB.
Improvement of Computational Model: In comparison to PLINDER-TIME and PLINDER-ECOD, the PLINDER-PL50 split exhibits the lowest amounts of leakage between training and test sets. Based on interactions, pocket location, and protein sequence similarity, the pocket_lddt graph efficiently eliminates connections. Though there is less leakage than with PLINDER-TIME, the PLINDER-ECOD split indicates problems with partial domain annotations. Due to the vast size of the PLINDER dataset, high diversity sets are guaranteed, and all PLINDER-PL50 test systems satisfy high-quality standards, providing trustworthy ground truth.
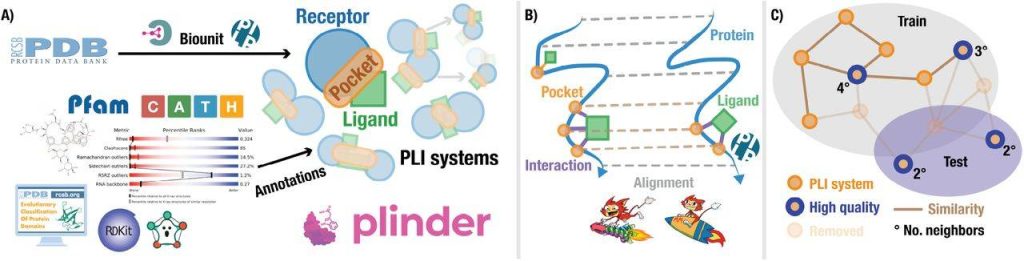
Image Source: https://doi.org/10.1101/2024.07.17.603955
Future Direction and Current Limitations
PLINDER is an all-inclusive resource designed to predict protein-ligand interactions and offers an extensive resource for techniques. More data annotations, including cross-docking scores, classical docking scores, and projected binding affinities, are being added to the AlphaFold Database, a platform used for training and testing models. In addition, several anticipated structures are linked to each system using the AlphaFold Database, increasing the variety of interactions, pockets, and folds covered. With an emphasis on smaller molecules and atom-level weighting for accuracy measures, alternative protein-ligand interaction profilers are being investigated to curate PLI systems. Per-residue Q-scores have been added to validation reports for electron microscopy structures; these might be included in criteria of a similar nature. A leaderboard with more realistic assessment scenarios of cross-docking or predicting postures inside apo and anticipated receptor architectures is planned. The techniques will be trained and validated on PLINDER datasets.
Conclusion
Researchers propose PLINDER, a vast, automatic, and comprehensive dataset repository for interactions between proteins and ligands. Researchers present the usefulness of scalable similarity metrics across protein-ligand complexes and a splitting approach that puts low information leakage and test set quality first. Researchers demonstrate that the approaches and findings offer a solid basis for dataset generation and quantify and control dataset leakage and quality in a tunable and quantitative way by retraining DiffDock on different splits.
Article Source: Reference Paper | The PLINDER source code is Apache 2.0 licensed and can be downloaded at GitHub.
Important Note: BioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}