
The complex molecular machinery that make up nature, proteins have evolved over billions of years and are essential to the continuation of life. However, a fundamental problem in contemporary biology is still interpreting their molecular language or comprehending how protein shapes and sequences encode and dictate biological functions. Here, scientists from Westlake University and Nankai University present Evolla, an 80-billion-parameter generative model designed to decode the molecular language of proteins. Evolla uses protein sequences and user queries to provide information about how proteins operate. It makes use of a dataset of 150 billion word tokens and 546 million protein question-answer pairs that were created by AI to comprehend the functional diversity and complexity of proteins. To improve response quality, Evolla integrates outside knowledge and adjusts its model in response to preference signals. Proteomics and functional genomics research is advanced by evaluating its effectiveness using a revolutionary paradigm called Instructional Response Space.
Introduction
Proteins are crucial molecular machinery that carry out an incredible variety of vital functions. Determining the molecular basis of life, discovering new drugs, and deciphering biological functions all depend on an understanding of protein function. Protein structure prediction was transformed by AlphaFold and its successors, which achieved previously unheard-of levels of accuracy. However, this development has brought attention to the discrepancy between structural determination and functional comprehension, which calls for new methods of functional annotation. This problem is made worse by the vast amount of contemporary protein data—less than half a million functional annotations have been expertly curated. A major obstacle in biological research and applications is the growing discrepancy between sequence and structural data and the lack of functional insights.
Finding similar proteins with known activities requires the use of structure comparison and protein sequence matching tools like Foldseek and BLAST. These techniques, however, are less useful for proteins without clear evolutionary relations because they mostly rely on similarity to previously identified proteins. The field of functional prediction has expanded due to recent developments in deep learning-based techniques, yet these methods frequently fail to produce comprehensive and context-specific insights. By figuring out the chemical “language” of proteins, large multimodal protein-language models present a promising way to reinterpret conventional protein function prediction challenges. Most datasets are restricted to 500,000 to 3 million protein-text pairs, with fewer than 100 million word tokens, which is much below the scale needed to depict the richness and complexity of protein biology adequately. Nevertheless, the training data utilized in these investigations continues to be a major bottleneck.
Looking into Evolla
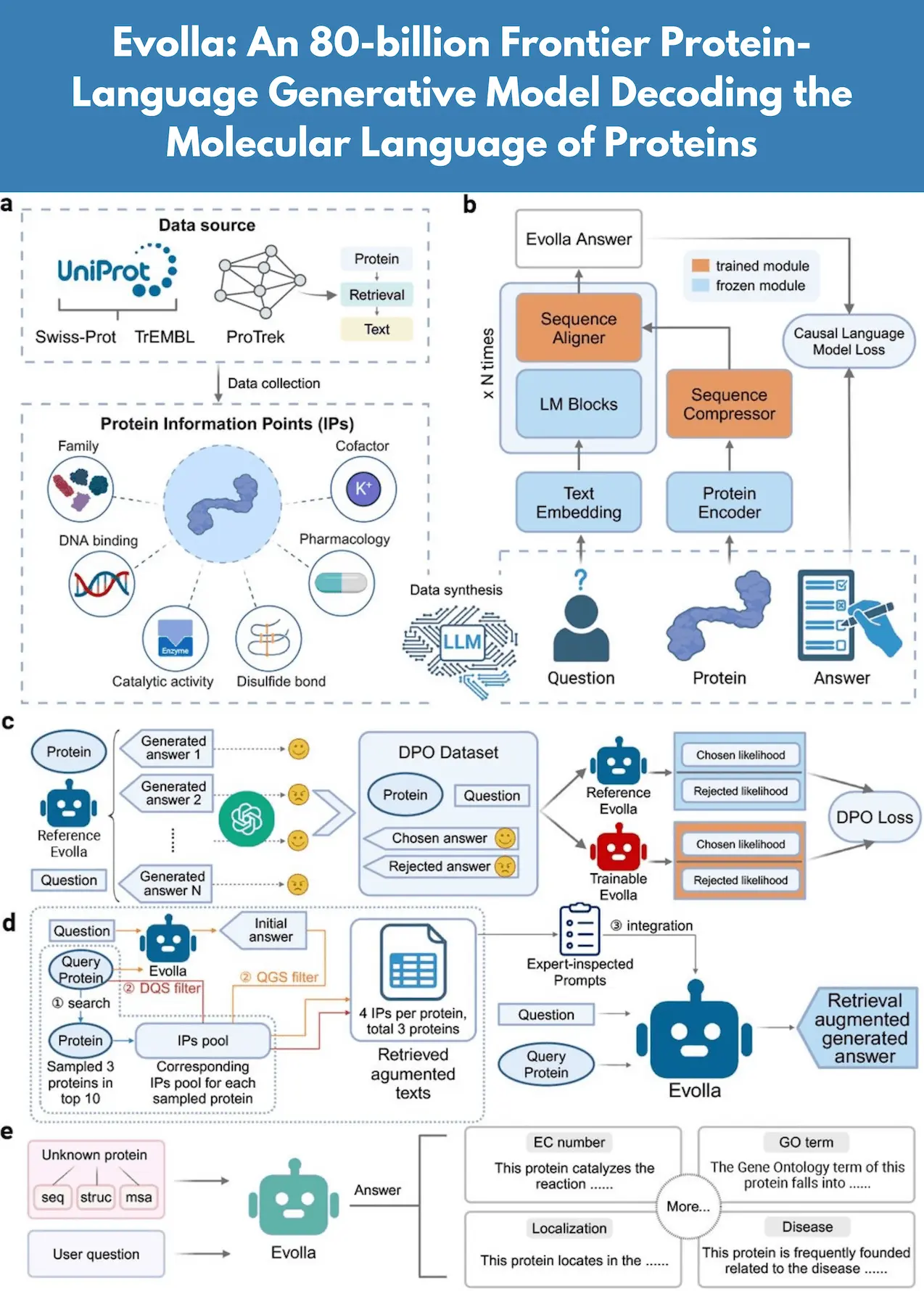
Researchers present Evolla, an 80B-parameter multimodal protein-language model that uses natural language conversation to understand protein chemical operations. With its unique architecture, Evolla combines three essential elements in a seamless manner: an intermediate compression and alignment module, a large language model (LLM) for decoding, and a protein language model (PLM) for encoding. An unparalleled scale in the field of protein-text modeling was achieved by training the model on a vast corpus of 546 million AI-generated protein-question-answer triples, which included 150 billion word tokens. An inventive pipeline that blends quick engineering, LLM distillation, and text-protein retrieval produced these large datasets by producing high-quality paired associations between proteins and their functional descriptions.
Evolla uses Direct Preference Optimization (DPO) to optimize output and Retrieval-Augmented Generation (RAG) to integrate knowledge and improve response quality. The extensive tests, which include a brand-new Instructional Response Space (IRS) assessment paradigm, show how well Evolla can produce accurate, complex, and contextually relevant protein functional responses. The capacity of Evolla to decipher the molecular language of proteins creates new opportunities for investigating the intricacy of life at a level and scope never before possible.
High-quality representations are extracted from a variety of protein data sources using Evolla’s protein encoder. A protein sequence is used by the Evolla model, an easy-to-use tool, to examine its characteristics and functions. It can be applied to illness association analysis, subcellular localization prediction, Gene Ontology annotation, and Enzyme Commission number classification. Because of its adaptability, the model is an effective tool for protein function annotation and more general biomedical research. It enables users to pose targeted queries and receive accurate, contextually relevant responses.
Conclusion
Evolla is a generative model of protein language with 80B parameters that improve the comprehension of protein molecular language. Using an AI-generated corpus of 546 million protein question-answer pairs, which totals 150 billion word tokens, it takes a novel method. This model produces accurate insights into protein function while preserving scientific accuracy thanks to the addition of Direct Preference Optimization and Retrieval-Augmented Generation. In advancing proteomics and functional genomics, Evolla creates a new paradigm for the large-scale interpretation of protein function by bridging the gap between protein sequences and structures. It also offers scientists a potent tool to solve basic riddles contained in protein molecules, expanding the comprehension of the molecular intricacy of life.
Article Source: Reference Paper | Evolla code repository is available on GitHub | The Evolla webserver is located at http://www.chat-protein.com/.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}