
In genomics, the conventional approach for explaining any gene expression is to cross-analyze bulk RNA sequencing (RNA-seq) data. However, as researchers, more often than not, start collaborating across different institutions, the problem of sharing sensitive information and keeping up with privacy concerns has become more apparent. To address this issue, Boris Muzellec, Ulysse Marteau-Ferey, and Tanguy Marchand from Owkin France developed FedPyDESeq2, the first system to allow DEA without sharing sensitive data. This proof of principle study across institutions shows the feasibility of the network of devices to deploy data-efficient solutions to enable privacy in genomic research.
The Problem: Centralization vs. Privacy
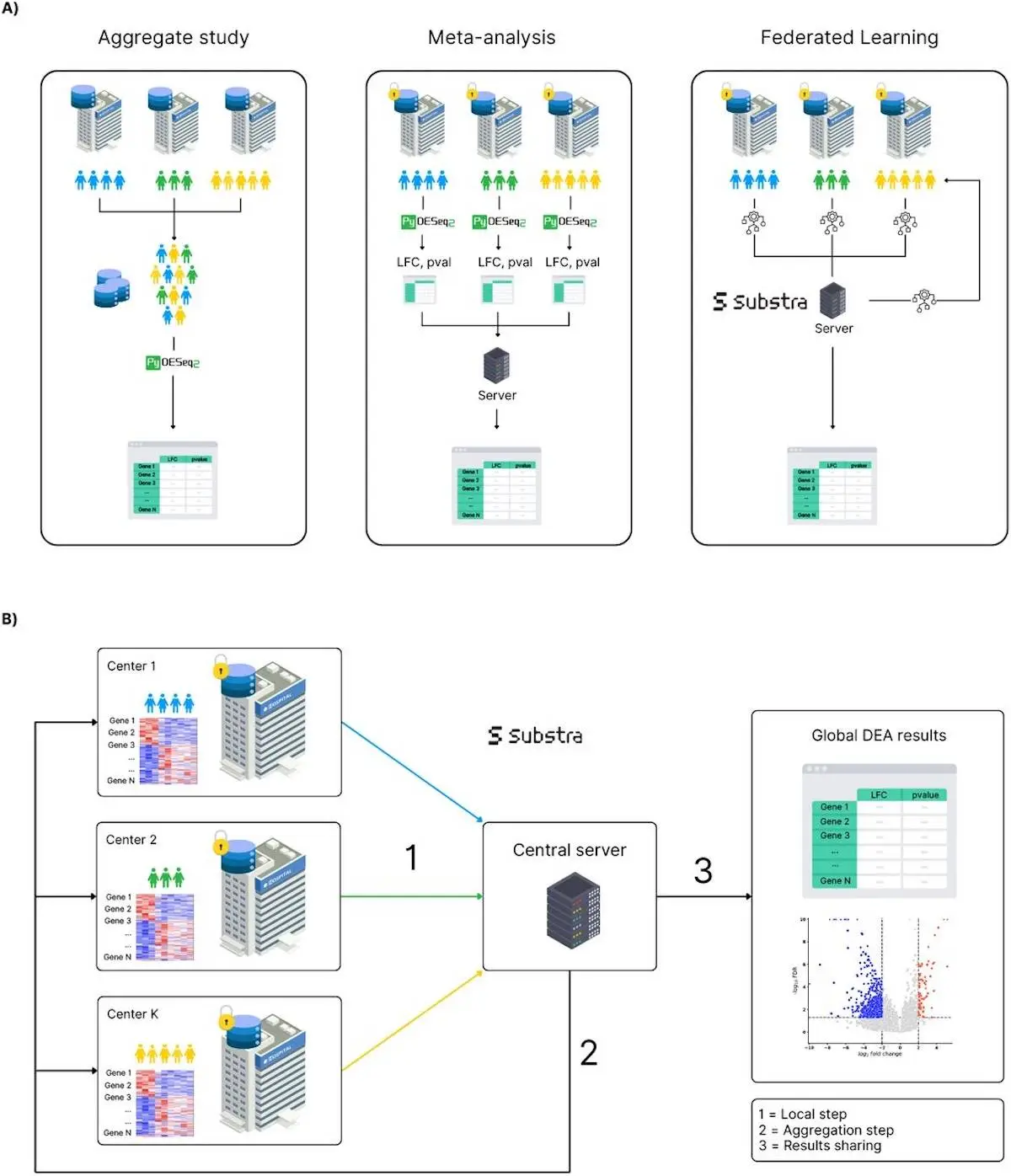
DESeq2 and other traditional tools are fast becoming the ideal approach in the bulk RNA-seq analysis continuum because they enable users to set conditions and withdraw statistically relevant genes that are differentially expressed under the set conditions. However, DESeq2 and similar tools require centralized data, where all gene count matrices and sample metadata are aggregated in one place. For studies that go through hospitals and biobanks or consortia, this type of centralization is often not achievable, mainly due to GDPR, HIPAA, and other ethical data protection regulations that are in place.
Decentralized data is a grave impediment to most policies. It is difficult to make a meaningful analysis without gathering sufficient statistics. This is where horizontal federation frameworks like FedPyDESeq2 become useful, as they allow collaboration while ensuring data is not compromised.
What is FedPyDESeq2?
FedPyDESeq2 is a modification of the well-established DESeq2 method tailored to work within the federated context. This framework makes it possible for several ‘centers’ to perform collaborative analysis of RNA-seq data through multiple protocols without the need to share specific personal data. Instead, it relies on the separable property of the negative log-likelihood (NLL) function that underpins the DESeq2 parameter optimization.
In a more simplified way, each center does its analysis with the data it has and only sends its findings to the central server, which is responsible for the final compilation. This server then combines these findings to refine the model to the desired parameters like dispersions and log-fold changes (LFCs) for every center. So, in effect, FedPyDESeq2 moves computation to the data, never sharing the raw data and therefore ensuring privacy while allowing for results that are on par with fully centralized approaches.
How it Works: Key Steps in FedPyDESeq2
- Size Factor Estimation
Each center computes size factors locally using the “median of ratios” method. The library size variability in the central ratios prevents the false sense of normality, which would result in biological gene expression differences being confused with technical ones. They then proceed to exchange their pseudo-reference values; this allows a uniform estimation of size factors across all entities.
- Initial Dispersion and Mean Estimates
To account for the levels of gene variability or gene heterogeneity, which is created through the utilization of a combination of local method of moments calculations and rough regression-based estimation. Each center’s data contributes to these initial values, which are later refined globally.
- Federated Gradient Descent
The core focus of this type of estimation is how the various models progressively fit the dispersions and the LFC using federated gradient descent. Each center local gradient is joined together, and an average is used to alter the core model parameters across all entities. This step simulates the core optimization of centralized DESeq2 but ensures the confines of data are always kept at the forefront.
- Outlier Detection and Adjustment
To narrow down the parameters that are to be included in the model counts, which might be disproportionately omitted from the analysis, If an outlier is detected and passes all the tests, the count is computed through a trimmed mean, and the newly computed parameters are then regressed.
- P-value Adjustment and Filtering
Statistical significance is assessed using Wald tests, with p-values adjusted using the Benjamini-Hochberg procedure. This ensures control of false discovery rates across genes. The final step—filtering genes based on outlier impact—further enhances the robustness of results.
Why FedPyDESeq2 Matters
FedPyDESeq2 is an important tool for collaborative RNA-seq analysis in the wake of numerous data protection coverages. It makes federated DEA possible and hence opens avenues for multi-institutional research on multiple conditions such as cancer, neurodegenerative disorders, and infectious diseases while maintaining data sovereignty.
The fact that the utility offers results that are not statistically different from those obtained using the centralized server utilizing DESeq2 means it has potential. This issue of loss of privacy or the guiding power of the analysis is eliminated by the use of FedPyDESeq2, which makes it a very suitable bioinformatics tool.
Conclusion
As more genomic applications adopt federated frameworks such as FedPyDESeq2, it is probable that new approaches will emerge, focusing on multi-genomic problems. For now, FedPyDESeq2 is a good answer to the contentious issue of protecting the privacy of the individual while allowing collaboration in the quest for new knowledge. It doesn’t matter whether you are a bioinformatician, clinician, or researcher; applications like these are helping create new pathways in the field of genomics in an interconnected manner without compromising on safety.
Article Source: Reference Paper | FedPyDESeq2 is available on GitHub under an MIT license | The scripts to reproduce experiments in the study are available here.
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}