
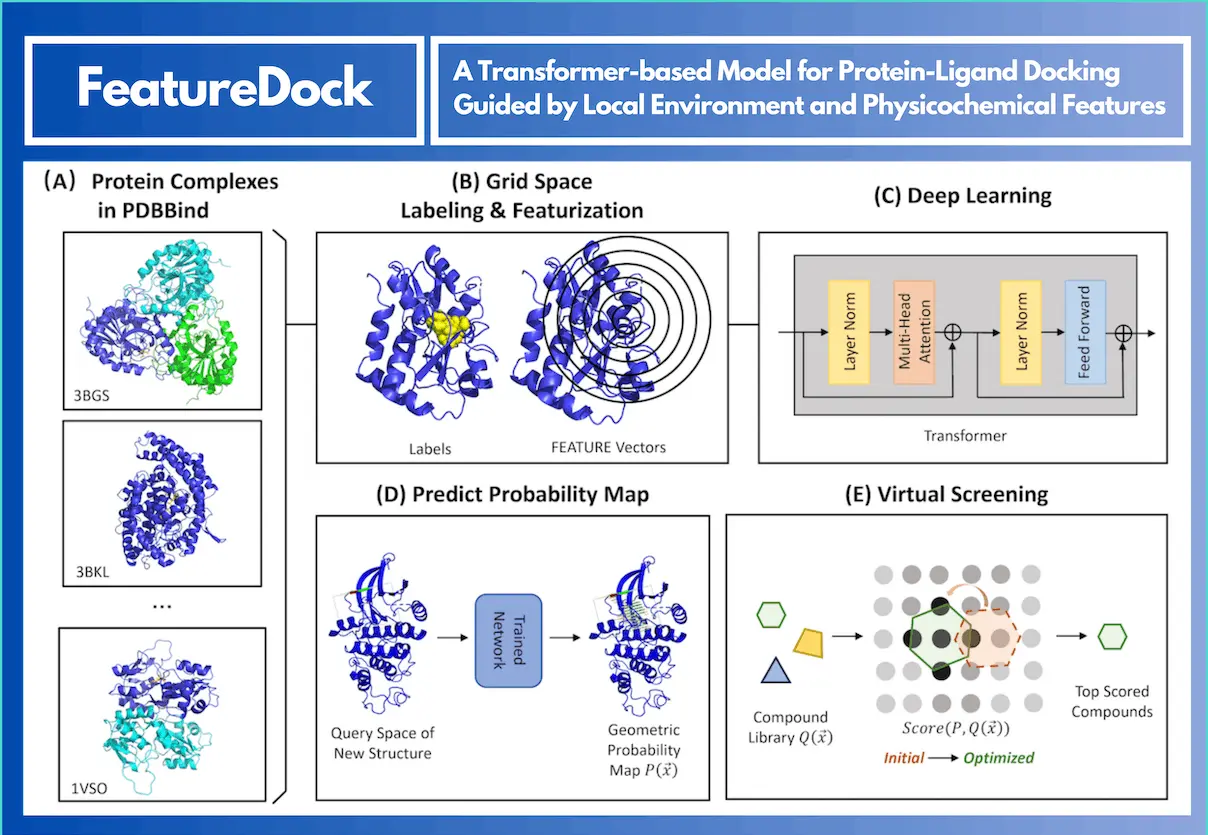
Molecular docking, which predicts the binding configurations between ligands and proteins, is an essential step in the drug discovery process. Advances in deep learning-based techniques such as DiffDock have demonstrated improved performance in binding pose prediction. Unfortunately, the lack of a clear scoring mechanism in these methods can make it difficult to distinguish between strong and weak inhibitors during virtual screening. To overcome this constraint, University of Wisconsin-Madison researchers provide FeatureDock. This transformer-based deep learning framework achieves a high scoring power for virtual screening in addition to properly predicting protein-ligand binding poses.FeatureDock is a model that extracts chemical features from local environments within protein structures to predict binding probabilities in the protein pocket. It uses a Transformer encoder to predict probability density envelopes, optimizing and scoring ligand poses. The model’s attention mechanism enhances interpretability by providing attention weights for each chemical feature.
Introduction
Pharmaceutical companies are often required to invest over ten years and billions of dollars in drug discovery before they can successfully introduce a new treatment to the market. It takes a long time for high-throughput screening (HTS) techniques to assess a compound library, which usually has millions of compounds. With just about 108 compounds now accessible in small molecule libraries like PubChem, ZINC, and ChEMBL, a number of in silico techniques have been developed to digitally screen possible compounds against the target to reduce the labor-intensive nature of HTS.
Evolution of Protein-Ligand Docking with Computational Tools
Employing docking-based techniques to forecast ligand binding locations, orientations, and conformations, AutoDock Vina is a potent tool in drug discovery. Conventional docking techniques, such as DOCK and AutoDock, rely on physics-based scoring functions to account for chemical interactions between and within molecules; however, these methods are computationally costly and frequently imprecise. Empirical scoring functions can overcome these restrictions by fitting parameters to experimental binding affinities using pre-selected descriptors, such as hydrophobic interactions and vdW-like potential. Smina has created an intuitive software interface that supports AutoDock Vina-based custom scoring functions. Additional AutoDock Vina variants have been designed to enhance the scoring function for particular systems, like halogen bond and carbohydrate systems.
Machine learning has been using neural networks more and more to increase scoring power through the usage of sophisticated topologies and extensive descriptions. The purpose of these networks is to forecast binding affinities in experiments; however, they are not applicable in situations where there are no 3D protein-ligand complexes. Protein-ligand complexes are treated as atom property tensors by 3D CNN models such as Atomic Convolutional Neural Networks (ACNN) and KDEEP. PotentialNet and other distance-aware graph neural networks (GNNs) encode Euclidean symmetries in structures.
Understanding FeatureDock
FeatureDock is a deep learning method that scores and poses small-molecule ligands binding to proteins using physicochemical properties. By extracting the local chemical environment for protein-RNA binding, 3D-invariant FEATURE representations are used to discretize possible binding pockets into grid points. Grid points in binding pockets with high probabilities for ligands to occupy generate probability density envelopes, which are predicted by the Transformer encoder. Transformer’s attention mechanism aids in illuminating and clarifying the significance of input characteristics. The ligands are screened and positioned in binding pockets using a position optimization technique and a unique scoring mechanism. FeatureDock is better at differentiating between strong and weak inhibitors than DiffDock, Smina, and AutoDock Vina, among other deep-learning models. The model accurately retrieves binding poses of top-predicted ligands, demonstrating a high level of proximity for CDK2.
Key Features of FeatureDock
- FeatureDock is a system that analyses the grid points encoded in the query pockets to predict and score the binding posture of protein complexes. In order to create probability density envelopes, a Transformer encoder model must be trained. The model then predicts the binding probabilities of these grid points. An algorithm for position optimization and a scoring function is then created using this data.
- Using the attention weights from the Transformer architecture, the FeatureDock program creates attention maps for each input chemical characteristic. Every attention map documents the role that a particular chemical characteristic plays in forecasting the binding probability of every grid point within the binding pocket.
- In the task for an inactivated version of CDK2 and the ACE receptor, the FeatureDock tool performs better than the classical docking approaches (Smina and AutoDock Vina) and the recently developed diffusion-based generative model (DiffDock).
- FeatureDock uses the chemicals’ alignment with the probability density envelopes to predict the binding poses and assess the binding potency of each component. Compared to other deep learning techniques like KDEEP and PotentialNet, which predict protein-ligand binding affinity values and rely on three-dimensional protein-ligand complexes as input, it, therefore, offers more information on the ligand poses and orientations.
Conclusions
Protein-ligand binding postures are predicted and scored using the feature-based Transformer framework, FeatureDock. It uses a transformer encoder to forecast probability density envelopes for specific protein pockets by extracting physicochemical data from the protein’s local environment. These anticipated envelopes establish the ligand’s preferred binding location at grid points inside the protein pockets. When compared to FNN and ResNet architectures, transformer encoders perform better, resulting in higher and more consistent prediction accuracy. By clarifying how each chemical property contributes to the final anticipated ligand binding probabilities, the encoder’s attention mechanism also has the potential to support rational drug design. In the virtual screening of drug-like compounds, FeatureDock performs better than DiffDock, Smina, and AutoDock-Vina, differentiating between strong and weak inhibitors and correctly predicting their binding poses.
Article Source: Reference Paper | FeatureDock is available on GitHub.
Important Note: ChemRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}